Büşra Kocameşe
Genel Bilgi
Abdominal Aort Anevrizmaları (AAA), aortun karın boşluğu kısmındaki bölümünde oluşan anevrizmalardır (damar genişlemesi). Her yaşta gerçekleşebilir ama en yaygın 50-80 yaş arası erkeklerde görülmektedir. Etkilenen çoğu birey belirti göstermezken bazılarında karın boşluğunda titreşim (nabız atması) hissi ve/veya sırt ağrısı görülebilir. Eğer anevrizma yırtılırsa çok şiddetli ağrıya, mide bulantısına, kusmaya, çarpıntıya ve şoka neden olur. AAA’ların %20’si er ya da geç yırtılır ve genelde ölümcüldür. Abdominal Aort Anevrizmalarında birden çok genetik ve çevresel faktör etkili olabilir. Bazen de kalıtsal bir sendroma bağlı olarak görülür. Bir ailede birden fazla bireyde bulunuyorsa “Ailesel Abdominal Aort Anevrizması” olarak adlandırılır.
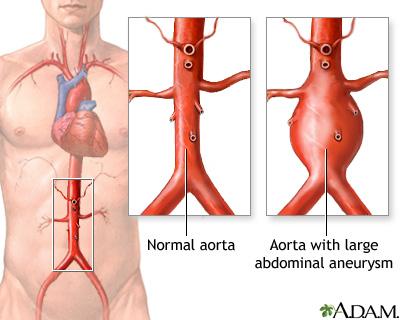
Kaynak: https://medlineplus.gov/ency/article/000162.htm
Belirtiler
Çoğu AAA hastasında hiç semptom görülmez ve tesadüfen başka bir amaçla yapılan karın bölgesi görüntülemesi (ultrason vb.) sırasında saptanır. Ya da fiziksel muayene sırasında abdomen stetoskopla dinlenirken anormal kan akışı sesi duyulabilir. Anevrizma genişlediği veya yırtıldığı zaman görülen en yaygın belirti, genelde karın bölgesinde başlayan ve sırta ve yanlara yayılan şiddetli ağrıdır. Diğer belirtiler anevrizmanın aortta nerede konumlandığına ve çevre yapıları etkileyip etkilemediğine bağlıdır. Yıllarca belirti göstermeden kalabilir.
Görülme Sıklığı
Abdominal aort anevrizması 60 yaş altı bireylerde nadiren görülür. 60-65 yaş arası 1000 kişiden birinde AAA görülür ve yaş arttıkça görülme sıklığı da artar. Tıbbi görüntüleme yöntemleri, AAA’ların 65 yaş üstü erkeklerin %2-13’sini; kadınlarınsa %6’sını etkilediğini göstermektedir. Ancak görüntüleme teknikleriyle belirlenen anevrizmaların neredeyse %90’ı küçük (çapı 3.5 santimden az) ve yırtılma ihtimali düşük olarak tanımlanmaktadır.
Genetik Değişiklikler
Abdominal Aort Anevrizması çok bileşenli (multifaktöriyel) bir rahatsızlıktır, yani genetik faktörler ve çevresel koşulların etkileşimiyle ortaya çıkar. Ailede AAA’ya sahip bireylerin bulunması hastalık riskini arttırır. İsveçli araştırmacılar, birinci derece yakınında AAA bulunan bir bireyde AAA gelişme ihtimalinin aile öyküsünde bu rahatsızlığın bulunmadığı bir bireye göre yaklaşık iki kat arttığını rapor etmişlerdir.
AAA kalıtsal sendromlara bağlı olarak da ortaya çıkabilir. Ehlers-Danlos Sendromu ve Marfan Sendromu, abdominal aort anevrizmasına neden olabilen, bağ doku bozukluğuyla karakterize iki hastalıktır.
Spesifik genetik değişimlerin AAA riskini arttırdığı tahmin edilse bile, sendroma bağlı olmayan AAA’ya sebep olan bir gen bilinmemektedir. Abdominal Aort Anevrizmasının kalıtımı karmaşık olduğu için bir bireyin bu rahatsızlığı geliştirip geliştirmeyeceğini öngörmek mümkün değildir.
Teşhis Yöntemleri
Fiziksel muayene AAA’yı teşhis etmek için birincil yöntemdir. Doktorun muayene sırasında karın boşluğunda pulsatil kitle hissetmesi ile teşhis konulabilir. Steteskopla dinleme sırasında da anevrizmadaki anormal kan akışı seslerinin duyulması mümkündür. Çoğunlukla ultrason vb. görüntüleme teknikleri ile AAA teşhis edilebilir ve anevrizmanın boyutu belirlenebilir.
Tedavi Yöntemleri
Abdominal Aort Anevrizması zaman geçtikçe genişler. Anevrizma büyüdükçe, yırtılması ve ölümle sonuçlanması riski de artar. Tedavi amacı anevrizma yırtılmadan önce müdahalede bulunmaktır. Semptom göstermeyen vakalarda anevrizmanın onarımının yapılıp yapılmayacağı ve ne zaman yapılacağının kararı risklere bağlı olarak değerlendirilir. Genelde çapı 4 santimden küçük olan anevrizmalarda hemen ameliyatla onarım yapılmasındansa zamanla kontrolünün yapılması önerilir. Çapı 5.5 santimden daha geniş olan veya 6 ayda 0.5 cm’den fazla genişleyen anevrizmalarda ise ameliyat önerilir.
Kaynakça








{kind=link}