Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
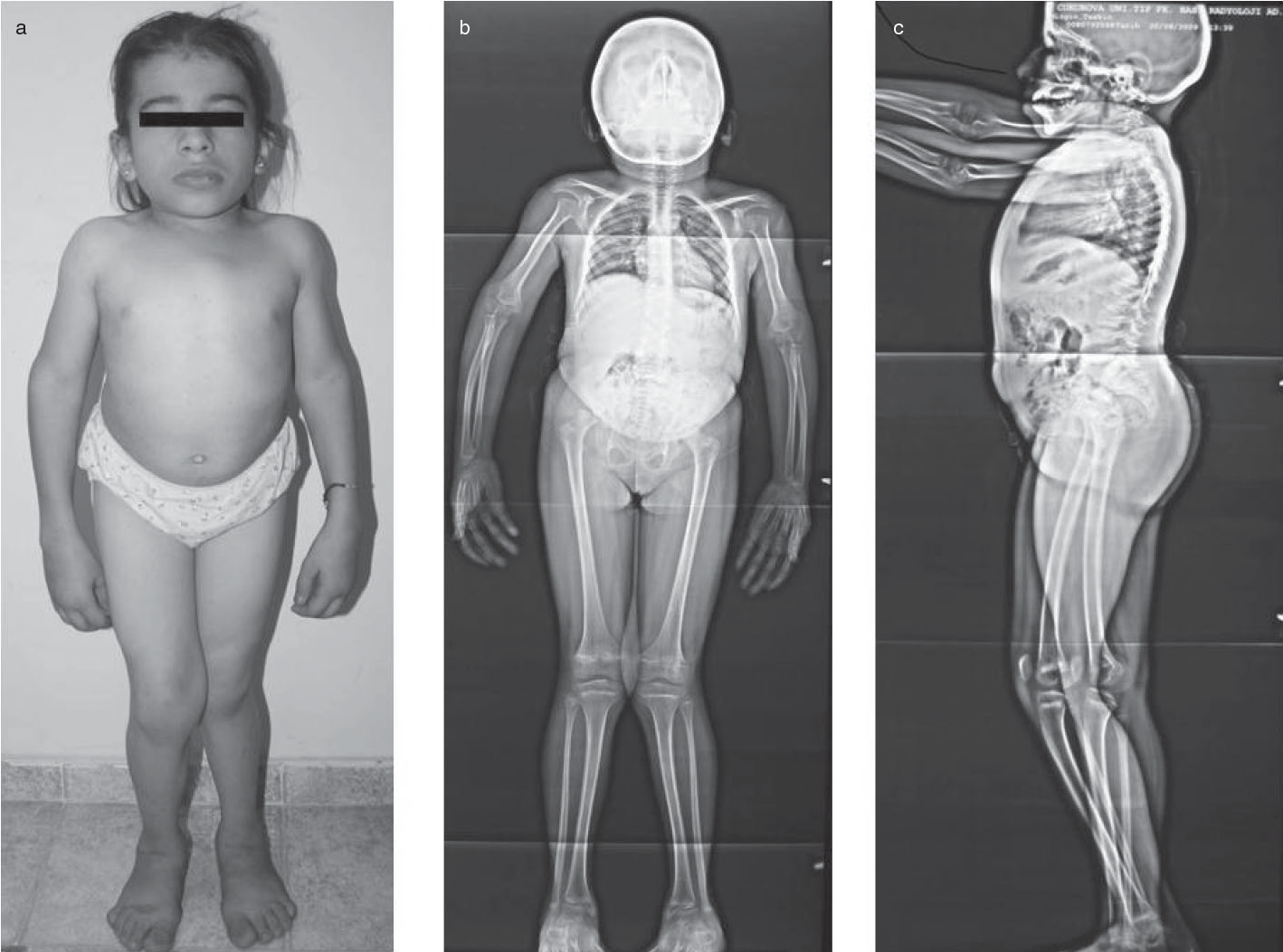
Arjininemi, arjinaz enzimi eksikliği sebebiyle üre döngüsünün gerçekleşmesini engelleyen genetik bir hastalıktır. Kromozom 6q23 üzerinde arjinaz enzimini kodlayan ARG1 geninde bulunan homozigot ya da çifte heterozigot mutasyon sebebiyle olur. Homozigot çekinik olarak bir sonraki nesile aktarılabilir. [1] Üre döngüsü vücudun metabolizmal aktivitesi sonucu üretilen azotun vücuttan atılması için gerekli olan bir döngüdür. Üre döngüsünün son enzimi olan arjinaz I arjinini, üre ve ornitine dönüştürür. [2] Bu enzimin düzgün çalışmaması sonucu kanda arjinin ve amonyak birikmesi olur. Genelde doğumda ve erken çocukluk döneminde semptomlar görülmez. 1 yaş ve 3 yaş arası tedavi edilmeyen bireylerde spastisite, bilişsel gelişimde bozukluk gelişir. Tedavi edilmemesi durumunda arjinaz enzimi eksikliğinde ciddi semptomlar görülür. [4]
Belirti ve Semptomlar
Arjininemi hastalığı bulunan bireylerde aşağıdaki belirti ve semptomlar görülebilir.
Anoreksi
Kusma
Zeka geriliği
Büyüme geriliği
Hiperamonemi
Hiperarjininemi
Spastik parapleji
Letarji
Nöbet
Atetoz
Bağırsak ve mesane kontrolü kaybı
Mikrosefali
Kortikal atrofi
Denge ve koordinasyon eksikliği
Genetik Görülme Sıklığı
1.100.000’de 1 ile 350.000’de 1 arasında görülme ihtimali olduğu tahmin edilmektedir.[4]
Kalıtım Paterni/Deseni:
6. kromozomda otozomal çekinik olarak aktarılır. Çerçeve kayması veya silinme mutasyonu sonucu oluşur. Otozomal çekinik olduğu için bireylerin %25 hasta, %50 taşıyıcı ve %25 ne hasta ne taşıyıcı olma ihtimali vardır. [4]
Teşhis Yöntemleri ve Tedaviler
Yeni doğmuş bebeklerde yapılan yeni doğan taramasında bu hastalığı fark etmek zordur. 1 yaşından sonra fark edilmeye başlanır. Plazmadaki arjinin seviyesinin normali 3-4 katına çıkması bu hastalığın olabileceğine dair işarettir. Kesin tanı moleküler genetik testler ile biallelik ARG1 patojenik varynatının testpiti veya kırmızı kan hücrelerinde arjinaz enzimi aktivetisinin olmaması ile konur. Üriner orotik asit konsantrasyonu bu bireylerde yüksek olabilir ama teşhis konması için birincil öncelikte değildir. [4] Çoğu üre döngüsü rahatsızlıklarına benzer bir tedavi yolu uygulanmaktadır. Bu hastalığa sahip olan bireylerde beslenme düzeninde değişiklik yapılarak dışarıdan protein alımının kısıtlanması ve temel amino asit ile desteklenip düşük proteinli beslenme ile tedavi edilebilir. Aynı zamanda intravenöz glukoz infüzyonu ile tedavi edilebilir. Akut durumlarda (koma ya da ensefalopatik) sodium benzoat ve/veya sodium fenilbütirat/fenilasetat kandaki amonyak seviyesini düşürmek için kullanılır. Nitrojen tutucu ilaçlar kullanılır.[3][5] Tedavilerden yanıt alınamadığında karaciğer naklide yapılabilmektedir. Valporik asitin bu hastalar üzerinde kullanılması önerilmemektedir. Kandaki arjinin seviyesinin kontrol edilmesi ve metabolik dengesinin takibi gerekmektedir. Eğer aile öyküsünde bu hastalığa sahip insanlar varsa yapılacak moleküler genetik testler ve plazma kantitatif aminoasit analizleri hastalığın erken teşhis ve tedavisinde çok önemli yer tutmaktadır.[4]
Hastalıkla İlişkili Genler
6p23’de bulunan arjinaz-1(ARG1) geninde bulunan homozigot ya da çifte heterozigot mutasyon sebebiyle olur. Çerçeve kayması, silinme ya da çoğalma mutasyonu ile oluşur. [1]
Hastalığın Diğer İsimleri
Arjininemi, arjinaz eksikliği, hiperarjininemi ve ARG1 eksikliği olarak da biliniyor.
Referans
[1] Cai, X., Yu, D., Xie, Y., & Zhou, H. (2018). Argininemia as a cause of severe chronic stunting and partial growth hormone deficiency (PGHD). Medicine, 97(7), e9880.doi:10.1097/md.0000000000009880
[2] Crombez E. A., Cederbaum S. D, (2004), Hyperargininemia due to liver arginase deficiency, doi:10.1016/j.ymgme.2004.11.004
[3] Fernando D. Testai., Philip B. Gorelick, (2010), Inherited Metabolic Disorders and Stroke Part 2: Homocystinuria, Organic Acidurias, and Urea Cycle Disorders Arch Neurol. 2010;67(2):148-153
[5] Qureshi, I. A., Letarte, J., Ouellet, R., Batshaw, M. L., Brusilow, S. Treatment of hyperargininemia with sodium benzoate and arginine-restricted diet. J. Pediat. 104: 473-476, 1984.
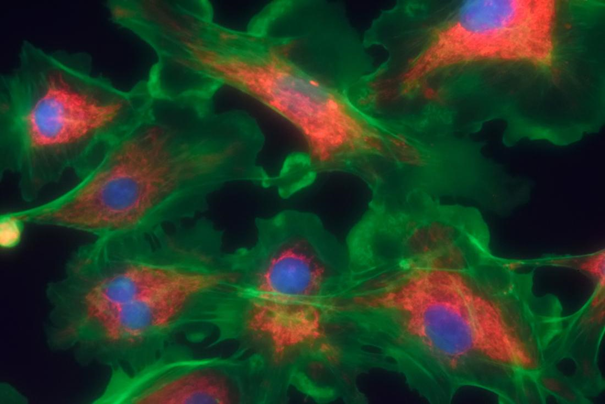
Fibrosarkom, kemik, kas ve diğer organları yerinde tutan fibröz doku hücrelerini. etkileyen mezenkimal kökenli nadir görülen bir tümördür. Vücut fibroblastların kontrolünü kaybeder ve fibroblastlar aşırı çoğalır. Bu durum gerekenden daha fazla lifli doku oluşturur. Bunun sonuucunda da tümör oluşumu gözlenir. Fibrosarkom da diğer kanser türleri gibi vücuda yayılabilir.(metastaz)
Figür 1: Fibrosarkomaya sahip fibroblast hücrelerinin 3D
görüntüsü
İki ana tip
vardır: Birincil Fibrosarkom ve ikincil Fibrosarkom.
Primer
fibrosarkom, değişken miktarlarda kolajen üreten bir fibroblastik malignitedir.
Sekonder fibrosarkom, önceden var olan bir lezyondan veya radyoterapiden sonra
kemik veya yumuşak doku alanına kadar ortaya çıkar. Bu daha agresif bir
tümördür ve prognozu daha kötüdür.
Nadiren,
fibrosarkom kemikte ortaya çıkabilir, ancak genellikle kemiğin kendisini değil,
yakındaki fibröz dokuyu etkiler.
Genetik Değişiklikler/Etken
Faktörler
Fibrosarkomun kesin nedeni
bilinmemektedir, ancak genetik mutasyonlar bir rol oynayabilir. Bazı nadir kalıtsal
hastalıklar insanlar da fibrosarkom görülme ihtimalini arttırır.Bu durumlar
şunları içerir:
Bu hastalıklara sahip bireylerde herhangi bir sarkom belirtisi olup
olmadığına dair düzenli kontrol edilmelidir.
Aynı zamanda;
zayıflamış veya hasar
görmüş lenf sistemi
radyasyona maruz kalma
muhtemelen vinil
klorür, arsenik ve dioksin dahil olmak üzere bazı kimyasal maruziyetlerde
Fibrosarkoma neden olabilir.
Belirti ve Semptomlar
Fibrosarkom en çok kollarda,
bacaklarda, göğüste veya karında görülür, ancak vücudun diğer bölgelerinde de
olabilir. Fibrosarkom belirtileri ilk başta hafif olabilir.Önceleri sadece cilt
altında ağrısız bir yumru veya şişlik görülebilir. Diğer belirtilerin
görüntülenmesi ise uzun zaman alabilir. Bu belirtiler şunları içerir.
Kol veya bacaklarda; tümör bölgesinde burkulma gibi hissedilebilecek
kalıcı ağrı görülmesi
Karında: Eğer Fibrosarkom
oluşumu karında
başlarsa, muhtemelen önemli boyutlara gelene kadar fark edilemez. İlerleyen
zamanlarda çevredeki organları, kasları, sinirleri veya kan damarlarını itmeye
başlayabilir. Bu durum acıya ve hassasiyete yol açabilir. Tümörün konumuna
bağlı olarak, solunum problemlerine yol açabilir. Ayrıca kabızlığa da sebep olabilir.
Mesanede bulunan
fibrosarkom alt karın bölgesinde ağrı, işemede zorlanma ve idrarda kan bulunmasına
sebep olur.
Baş veya boyunda gözlenen
Fibrosarkomda ise belirtiler burunda tıkanıklık ve burun veya boğazdan akıntı
şeklinde görülür. Bazı durumlarda göz şişebilir.
Bunların yanında aşağıdakiler gibi başka semptomlar da
gözlemlenebilir.
yorgunluk
iştah kaybı
kilo kaybı.
eklem veya uzuvu hareket ettirmede zorluk
tümörün sinirlere baskı yapması nedeniyle vücudun ilgili
bölgelerindeki uyuşma
Ancak semptomları başka sarkom türleriyle de
benzerlik gösterdiği için Fibrosarkomun sadece semptomlarla tanımlanması kolay
değildir.
Genetik Görülme Sıklığı
Fibrosarkom, kas-iskelet
sarkomlarının sadece % 10’unu ve tüm primer kemik tümörlerinin% 5’inden daha
azını temsil eder. Nadirdir, her 2 milyon
kişiden birini etkiler. Herhangi bir yaştaki
hastalarda teşhis edilebilir, ancak yaşamın dördüncü dekadındaki hastalarda
daha sık teşhis edilir.
Kalıtım Paterni/Deseni
Fibrosarkomun kalıtım paterni bilinmemektedir. Kemik fibrosarkomu erkeklerde kadınlardan biraz daha sık görülür.Bilinen ırksal tercih yoktur.
Teşhis
Yöntemleri ve Tedaviler
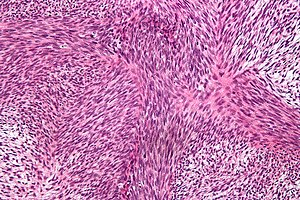
Figür 2: Fibrosarkomun resmi balıksırtı deseni
olarak tanımlamır. Bu model çok belirgindir ve genellikle fibrosarkom tanısını
doğrular.
Teşhis için öncelikle görüntüleme
yöntemleri kullanılır. Bu yöntemler tümörleri ve diğer anormallikleri
tanımlamayı kolaylaştıran ayrıntılı resimler üretir. Bir kitle bulunursa,
fibrosarkomu doğrulamak adına biyopsi yapılır. Biyopsi sonrası bir patolog
tarafından, herhangi bir kanser hücresi olup olmadığını ve varsa ne tür olduklarını
belirlemek için örnekler analiz edilir. Eğer hastalık varsa, tümör
derecelendirilir. Fibrosarkom tümörleri 1 ila 3 ölçeğinde derecelendirilir.
Kanser hücreleri, normal hücrelere ne kadar az benzerse, derece o kadar yüksek
olur. Yüksek derecelendirilen tümörler diğerlerine nazaran daha agresif olma
eğilimindedir. Yani daha hızlı yayılırlar ve tedavileri daha zordur.
Doktorlar, tedavi planını
aşağıdakiler gibi birçok faktöre dayandıracaktır:
primer tümörün derecesi, boyutu ve
yeri
kanserin yayılıp yayılmadığı ve ne
kadar yayıldığı
yaşınız ve genel sağlığınız
bunun önceki bir kanserin nüksü olup
olmadığı
Fibrosarkom için ana tedavi, tümörün
çıkarıldığından emin olmak için tümörün etrafında geniş kenar boşlukları ile
primer tümörü çıkarılmasına dayanır. Bazı durumlarda amputasyon gerekebilir. Radyasyon
tedavisi ve kemoterapi gibi yardımcı tedavi lokal kontrolü iyileştirebilir ve
klinik olarak belirgin metastatik hastalığın ortaya çıkma olasılığını
azaltabilir. Tedavi sonrasında da hastalar genellikle en az 5 yıl boyunca
izlenir.
Hastalıkla
İlişkili Genler
Genetik mutasyonlar rol
oynayabilir. Mevcut araştırmalar birçok sarkomun bu mutasyonlarla ilişkili
olduğunu göstermektedir. Ancak hastalık sebebi ve ilgili genler henüz
bilinmemektedir.
Burkitt lenfoma, ağırlıklı olarak Orta Afrika’daki
küçük çocukları etkileyen nadir bir kanser türüdür, ancak hastalık diğer
bölgelerde de bildirilmiştir. Afrika’da görülen örneklerde, patojenik mekanizma
belirsiz olmasına rağmen, Epstein-Barr virüsü enfeksiyonu ile ilişkili olduğu
tahmin edilmektedir.
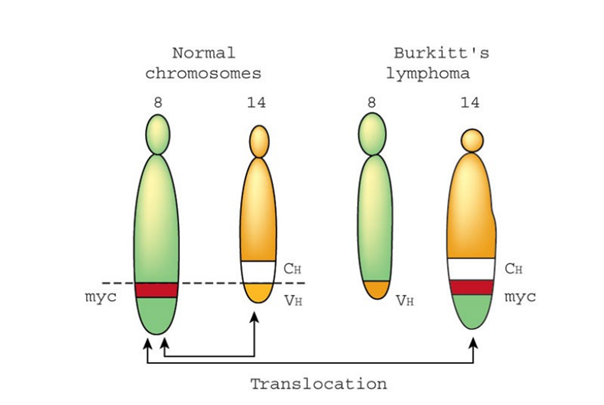
Burkitt lenfoma Myc genindeki somatik mutasyondan
kaynaklanır ve Myc geni ve immünoglobulin genleri ile beraber translokasyonlar
söz konusudur. Karşılıklı kromozomal translokasyonlar, lösemi ve lenfoma gibi
beyaz kan hücrelerini kapsayan iki tip kanserin de aralarında bulunduğu birçok
kanserin karakteristik özelliğidir.
Burkitt lenfoma hastalarından alınan kan örneklerinde
kromozom analizi yapılırken kromozom 8 (c-myc geninde ya da yakınında olan
translokasyon kırılma noktası ile) ve kromozom 2, 14 veya 22 (immünoglobulin
genlerinden birisinin yanında ya da üzerinde olan translokasyon kırılma noktası
ile) aralarında karşılıklı translokasyonlar görülmektedir. Burkitt lenfoma
hastasının c-myc geninde ve immunoglobulin genlerinin DNA sekanslamasında eşsiz
translokasyon kırıklıkları meydana gelir.
Bu hastalarda eşsiz bir şekilde translokasyon
kırıklıkları vardır bu da her Burkitt lenfoma vakasında kanser hücrelerinin
tamamının tek hücreden oluştuğunu ve bu atasal hücrenin de genetik
bozukluklarını nesillerine aktardığı gözlenir.
Kromozom translokasyonuna neyin sebep olduğu hala kesin olarak bilinmemektedir. Bununla birlikte, fareler gibi model organizmalarda yapılan araştırmalar, translokasyonların nasıl meydana geldiğini ve bu sürecin Burkitt lenfoma ve lösemi gibi diğer kanserlere nasıl katkıda bulunduğunu daha iyi anlamamıza yol açmaktadır.
Viral hemorajik ateş, son zamanlarda keşfedilen, şiddetli,
çoklu ve sıklıkla ölümcül kanamalarla karakterize ,bulaşıcı viral
enfeksiyonların bir grubudur. Afrika ateşleri arasında 1969’da keşfedilen Lassa
ateşi, ilk olarak 1967’de ortaya çıkan Marburg hastalığı ve 1976’da ortaya
çıkan Ebola ateşi bulunur. Diğer virüsler de hemorajik ateşlere neden olabilir .
Lassa humması ile ilgili olarak, 7
günlük bir kuluçka döneminden sonra, ateş ve ülseratif hemorajik farenjit,
ardından akciğerlerle birlikte göğüs zarının yangısı meydana gelir.
Hastalık daha sonra vakaların % 35 ila %70’inde ölüme yol
açan sindirim veya akciğer kanamalarına neden olur. Ebola’nın insanlar arasında
bulaşması, hava kaynaklı yoldan ziyade esas olarak hastalıkla veya enfekte
biyolojik ürünlerle doğrudan temas yoluyla gerçekleşir. Klinik bulgular 4-16
günlük bir kuluçka döneminden sonra, başlangıçta ateş, baş ağrısı, kas ağrısı
ve göz kızarıklığı ile ortaya çıkar. Daha sonra mide bulantısı, kusma ve ishal
gibi sindirim semptomları ortaya çıkar. Bir sonraki aşama ise; burun, bağırsak veya genital bölgede kanamanın
ortaya çıkmasıdır.Hastalık genellikle birkaç gün içinde ölümcüldür.
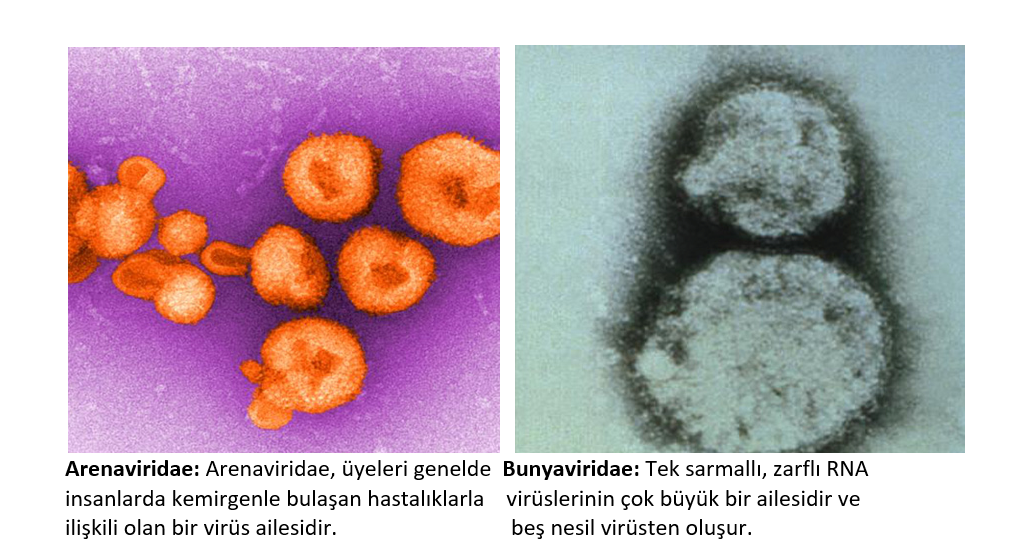
Beş RNA
virüsü ailesinin hemorajik ateşe neden olabildiği bulundu.
Arenaviridae ailesi arasında; Lassa humması (Lassa
virüsü), Lujo virüsü, Arjantin (Junin virüsü), Bolivya (Machupo virüsü),
Brezilya (Sabiá virüsü), Chapare hemorajik ateşi (Chapare virüsü), Venezüella
(Guanarito virüsü) ve Whitewater Arroyo virüsü bulunur.
Bunyaviridae ailesi, böbrek sendromlu Hantavirüs
hemorajik ateşinin (HV-HFRS) , Orthobunyavirus ve Rift Vadisi ateşinden (RVF) gelen
Kırım-Kongo kanamalı ateşinin (CCHF) virüsünü içerir.
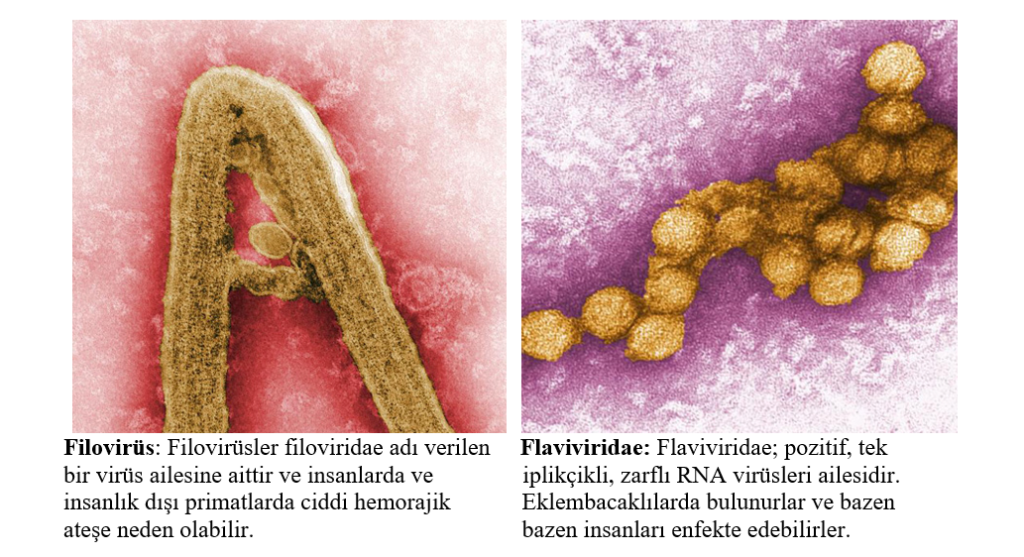
Filoviridae familyasında Ebola virüsü ve Marburg
virüsü bulunur.
Flaviviridae ailesi; kene kaynaklı ensefalit
grubunda VHF’ye neden olan dengue, sarı
humma ve iki virüsü içerir. Bunlar Omsk hemorajik ateş virüsü ve Kyasanur Orman
Hastalığı virüsü.
Eylül 2012’de PLOS Pathogens
dergisinde yazan bilim adamları, Demokratik Kongo Cumhuriyeti’nin Bas-Kongo
bölgesinde 2 ölümcül ve 2 ölümcül olmayan hemorajik ateş vakasından sorumlu bir
Rhabdoviridae üyesinin izole edildiğini bildirdi. Virüs, Bas-Kongo virüsü
olarak adlandırıldı.
Kesin tanı
genellikle ileri biyo-koruma yeteneklerine sahip laboratuvarlarda yapılır.
Laboratuvar
araştırması bulguları virüsler arasında bir miktar farklılık gösterir, ancak
genel olarak toplam
beyaz hücre sayısında (özellikle lenfositler) bir azalma, trombosit sayısında
bir azalma, kan serumu karaciğer
enzimlerinde bir artış ve kan pıhtılaşma yeteneğinde azalma vardır.
Hem protrombin (PT)
hem de aktive parsiyel tromboplastin sürelerinde (PTT) bir artış olarak
ölçülmüştür.Hematokrit yükselebilir. Üre ve kreatin yükseltilebilir, ancak bu
hastanın hidrasyon durumuna bağlıdır. Kanama süresi uzama eğilimindedir.
Önlem
Sarıhumma aşısı haricinde ne aşılar ne
de deneysel aşılar kolayca elde edilebilir. Profilaktik (önleyici) ribavirin,
bazı bunyavirüs ve arenavirüs enfeksiyonları için etkili olabilir (sadece yeni
bir araştırma ilacı (IND) olarak mevcuttur).
VHF izolasyon yönergeleri, tüm VHF
hastalarının (dang hastaları hariç) el hijyeni, çift eldivenler, önlükler,
ayakkabı ve bacak kaplamaları ve yüz koruyucu veya gözlükler dahil olmak üzere
sıkı temas önlemlerini kullanmaları gerektiğini belirtir.
Lassa, CCHF, Ebola ve Marburg
virüsleri özellikle hastane kaynaklı yayılmaya eğilimli olabilir. En azından,
uygun test edilmiş, HEPA filtre donanımlı bir solunum cihazı (N-95 maskesi
gibi), pille çalışan, hava temizleyici bir solunum cihazı veya pozitif bir
basınç beslemeli hava maskesi dahil olmak üzere hava yoluyla önlemler alınmalıdır.
VHF hastasının 1,8 metre yakınına gelen personel tarafından giyilir.Hasta
grupları ayrı bir binaya veya izole bir klima santraline sahip bir koğuşa
koyulmaldır.
Çevresel dekontaminasyon tipik olarak
hipoklorit (örn. Ağartıcı) veya fenolik dezenfektanlarla gerçekleştirilir.
VHF hastalarının tıbbi tedavisi yoğun
destekleyici bakım gerektirebilir. İntravenöz ribavirin ile antiviral tedavi
Bunyaviridae ve Arenaviridae enfeksiyonlarında (özellikle Eski Dünya Hantavirüs
enfeksiyonu nedeniyle Lassa ateşi, RVF, CCHF ve HFRS) faydalı olabilir ve
sadece ABD Gıda ve İlaç İdaresi tarafından onaylanan IND olarak deneysel bir
protokol altında kullanılabilir . İnterferon Arjantin veya Bolivya hemorajik
ateşlerinde etkili olabilir (sadece IND olarak da mevcuttur).
Philadelphia, PA, ABD’de 1793 Büyük Sarı Ateş Salgını.
50.000 nüfusun yaklaşık% 10’u hastalığa yenik düştü.
Gabon’daki Mékambo, Ebola virüsü hastalığının çeşitli
salgınlarının bulunduğu yerdir.
Orientale Eyaleti, Durba ve Watsa’nın Kongo Demokratik
Cumhuriyeti, 1998-2000 Marburg virüsü hastalığı salgınının merkeziydi.
Angola’daki Uíge Eyaleti, 2005 yılında bu hastalığın
bugüne kadarki en büyük Marburg virüsü hastalığı salgınının yapıldığı yerdi.
Kongo Demokratik Cumhuriyeti Mweka köyünde Ağustos
2007’de başlayan ve 103 kişiyi (100 yetişkin ve üç çocuk) öldüren bir VHF
salgınının (en azından kısmen) Ebola virüsü tarafından kaynaklandığı
gösterilmiştir.
Viral hemorajik ateş, Peloponezya Savaşı sırasında Atina
Vebasının olası bir nedenidir.
Viral hemorajik ateş, Kara Ölüm’ün ve Justinian Veba’nın
nedeninin alternatif bir teorisidir .
Eylül-Ekim 2008’de Lujo virüsünün ilk ve şu anki salgını
4/5 hastayı öldürdü.
Tarihin en büyük salgını olan 2014 Batı Afrika Ebola
salgınıdır.
Daha yaygın
olarak ARSACS olarak bilinen Charlevoix-Saguenay’ın otozomal resesif spastik
ataksisi, kas hareketini etkileyen bir durumdur. ARSACS hastalarında tipik
olarak kasların anormal gerilmesi , denge ve koordinasyon sorunları ve kol ve
bacaklarda duyu ve güçsüzlük) vardır. ARSACS’ta ortaya çıkabilecek ek
problemler arasında kas kaybı, istemsiz göz hareketleri ve yutma güçlüğüve
konuşma bulunur. ARSACS’ın diğer özellikleri arasında yüksek kemerli ayaklar,
yana eğilen bir omurga, gözün arkasındaki ışığa duyarlı dokuda sarı yağ dokusu
çizgileri , idrar sistem sorunları, zihinsel engel, işitme kaybı ve tekrarlayan
nöbetler (epilepsi). Dengesiz bir yürüyüş tarzı ARSACS’ın ilk belirtisidir.
Yürüme sorunları genellikle yürümeye başlayan çocuklar yürümeyi
öğrendiklerinden 12 ay ile 18 ay arasında başlar. Bu hareket problemleri
zamanla kötüleşir, spastisite (kasların şiddetli
derecede kasılı kalması hali) ve kol ve
bacak ataksisi (denge bozuluğu) artar. Bazı durumlarda spastisite gider, ancak
bu belirgin iyileşmenin kol ve bacaklardaki sinirlerin boşa gitmesinden
kaynaklandığı düşünülmektedir. Etkilenen bireylerin çoğu otuzlu veya kırklı
yaşlarında tekerlekli sandalye yardımına ihtiyaç duyar.
Hastalık ismini
ilk görüldüğü alandan almakta (Kanada’nın Quebec kentindeki Charlevoix-Saguenay
bölgesi). ARSACS dünya çapında bireylerde tanımlanmıştır.
Görülme Sıklığı
Charlevoix-Saguenay
bölgesindeki ARSACS insidansının 1.500 ila 2.000 kişide 1 olduğu tahmin
edilmektedir. Quebec dışında ARSACS sıklığı bilinmemektedir. Bilimsel
literatürde ARSACS’li yaklaşık 200 kişi tanımlanmıştır.
Hastalığın Nedeni
SACS genindeki
mutasyonlar ARSACS’a neden olur. SACS geni, saksin adı verilen bir protein
üretmek için talimatlar sağlar. Sacsin öncelikle beyin, cilt ve hareket için
kullanılan kaslarda (iskelet kasları) hücrelerde bulunur, ancak proteinin
spesifik işlevi bilinmemektedir.
Aspartilglukosaminüri , merkezi sinir
sistemini kapsayan ve iskelet anormalliklerine ve bağ dokusu lezyonlarına neden
olan ciddi otozomal resesif lizozomal depo bozukluğudur. En karakteristik
özelliği ilerleyici zihinsel geriliktir. Bozukluğa, lizozomal enzim
glikosilasparaginazın yetersiz aktivitesi neden olur; AGU, genellikle
Finlandiya hastalık mirası olarak adlandırılan bozukluklar grubuna aittir.
Belirtiler
Aspartilglikozaminüri olan bebekler doğumda
sağlıklı görünür ve gelişim çocukluk boyunca tipik olarak normaldir. 2 veya 3
yaşlarında ortaya çıkan bu durumun ilk işareti genellikle konuşma gecikmesidir.
Hafif zihinsel engelli belirtisi daha sonra belirginleşir ve öğrenme yavaş bir
hızda gerçekleşir. Entelektüel sakatlık ergenlik döneminde giderek kötüleşir.
Bu bozukluğu olan çoğu insan öğrendikleri konuşma yeteneğinin çoğunu kaybeder
ve etkilenen yetişkinlerin kelime dağarcığında genellikle sadece birkaç kelime
vardır. Aspartilglukozaminüri olan yetişkinlerde nöbet veya hareketle ilgili
sorunlar gelişebilir. Bu durumdaki insanlarda giderek zayıflayan ve kırılmaya
müsait hale gelen kemikler
(olabilirosteoporoz), alışılmadık derecede geniş bir eklem hareketi (
hipermobilite ) ve gevşek cilt semptomları görülür. Etkilenen bireyler, geniş
aralıklı gözler ( oküler hipertelorizm ), küçük kulaklar ve dolgun dudaklar
içeren karakteristik bir yüz görünümüne sahiptir . Burun kısa ve geniştir ve
yüz genellikle kare şeklindedir. Bu duruma sahip çocuklar yaşları için uzun
olabilir, ancak ergenlik döneminde büyüme hamlesi olmaması tipik olarak
yetişkinlerin kısa olmasına neden olur. Etkilenen çocuklar da sık sık üst
solunum yolu enfeksiyonu geçirme eğilimindedir. Aspartilglukozaminüri olan
bireyler genellikle yetişkinliğin ortalarına kadar hayatta kalırlar. İskelet
deforme olabilir. Omurga bükülmüş olabilir (skolyoz) ve boyun alışılmadık
derecede kısa olabilir. Gözler de katarakt gelişebilir. Davranış sorunları
yaygındır. Akciğer, kalp ve kan problemleri sonraki yıllarda ortaya çıkma
eğilimindedir. Bireylerde %80-%90 görülen belirtiler;
amino asit metabolizmasında anormallik
idrarda yüksek aspartilglukozamin seviyeleri
Gecikmiş konuşma ve dil gelişimi
İstemsiz kas hareketleri bozukluğu
Dişeti büyümesi
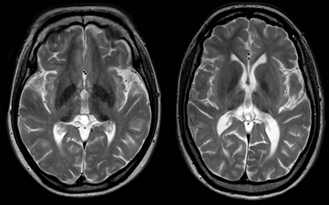
[1.1] İki aspartilglikozaminüri hastası 3.0 T olarak görüntülendi. Solda 34 yaşında kadın hasta, sağda 26 yaşında kadın hasta. Pulvinar çekirdeklerde tipik sinyal yoğunluğu azalması gösteren eksenel T2 ağırlıklı görüntüler. Her ikisinde de, gri ve beyaz madde arasındaki zayıf farklılaşma, sinyal yoğunluğu artışı olan bazı düzensiz joktakortikal odaklar, 3. ventrikülün hafif dilatasyonu, bir epifiz kisti ve nispeten kalın bir kafatası da görülebilir.
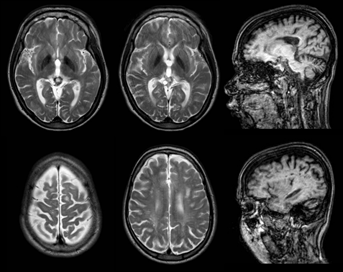
[1.2] 33 yaşında kadın aspartilglikozaminüri hastası, 3.0 T’de görüntülendi. Solda, daha önce aspartilglikozaminüri ‘da bildirilmeyen talamide atipik sinyal yoğunluğu artışı gösteren dört eksenel T2 ağırlıklı görüntü ve periventriküler ve juktakortikal beyaz maddede sinyal yoğunluğu artışı olan birkaç düzensiz odak var. Sağda pulvinar çekirdeğindeki SI azalmasını ve beyaz maddedeki bazı fokal hipertansiyon lezyonlarını gösteren iki sagital T1 ağırlıklı görüntü vardır. Ayrıca bu hastanın görüntülerinde hafif lateral ve 3. ventrikül dilatasyonu, gri ve beyaz madde arasındaki zayıf farklılaşma, hafif dilate perivasküler boşluklar, pineal kist ve hafif genel atrofi görülebilir.
Nedenler
AGA genindeki mutasyonlar aspartilglukosaminüriye
neden olur. AGA geni, aspartylglucosaminidase olarak adlandırılan bir enzim
üretmek için talimatlar içerir. Bu enzim, geri dönüşüm merkezleri olarak işlev
gören hücrelerdeki lizozomlarda aktiftir. Lizozomlar içindeki enzim, belirli
proteinlere (glikoproteinler) bağlı şeker molekülleri (oligosakkaritler)
komplekslerinin parçalanmasına yardımcı olur. AGA gen mutasyonları,
lizozomlarda aspartilglukosaminidaz enziminin yokluğuna veya eksikliğine neden
olarak glikoproteinlerin normal parçalanmasını önler. Sonuç olarak, lizozomlar
içinde glikoproteinler birikebilir. Fazla glikoproteinler hücrenin normal
fonksiyonlarını bozar ve hücrenin tahrip olmasına neden olabilir. Bir
glikoprotein birikimi özellikle beyindeki sinir hücrelerini etkilemektedir; bu
hücrelerin kaybı aspartilglukosaminüri belirtilerinin ve semptomlarının çoğuna
neden olur .
Epidemiyolojisi
Aspartilglikozaminüri ‘nın Finlandiya’daki 18.500 kişiden 1’ini etkilediği tahmin edilmektedir. Bu durum Finlandiya dışında daha az yaygındır, ancak oranı bilinmemektedir.
Kalıtım Kalıbı
Bu durum otozomal resesif paternde kalıtsaldır
yani her hücredeki genin her iki kopyasının mutasyonları vardır. Otozomal
resesif koşulu olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin
bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını
göstermezler.
Teşhis Yöntemleri
Biyokimyasal olarak, bir amino asit veya oligosakkarit
kromatografisi üzerine aspartilglikozaminin çokça bulunduğu idrarın atılımı ile
karakterizedir. Sonuçlar, lenfositler, fibroblastlar, amniyositler veya
trofoblastta ölçülen düşük aspartilglikozaminidaz aktivitesi ile doğrulanır.
Tedavi
Bu güne kadar tedavi edici tek girişim, 5 Fin
hastası ile sınırlı allojenik kemik iliği aşılama işlemiydi.
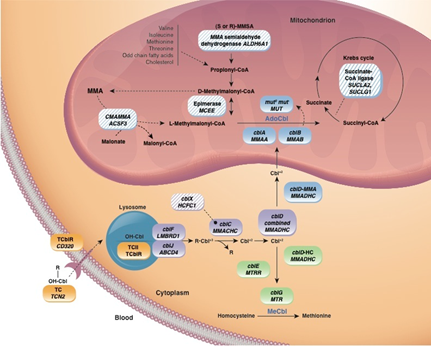
B12
bağımlı bir enzim olan metilmalonil-KoA mutaz enziminin eksikliğinin yol açtığı
otozomal resesif geçişli bir metabolik hastalıktır. Bu enzim
Metilmalonil-KoA’nın süksinilKoA’ya dönüşümünü sağlaryani bu enzim yoksa
metilmalonil-KoA süksinilKoA. ya dönüşemeyeceğinden dolayı birikir kanda
düzeyi çok artınca da idrarda metilmalonik asit atılımı gerçekleşir.
Hastalık kalıtsal olup otozamal resesif genler ile taşınmaktadır.
Belirti ve Semptomlar
Belirtiler yeni doğan
döneminden erişkin dönemine kadar devam eder. Yaşamın çok erken döneminde 1 ila
4 hafta arasında bu belirtiler anında kendisini gösterir. Genel olarak hastalık kendiliğini yaşamın ilk
anlarından itibaren kusma , gelişmede gerilik , kas hipotoni ,katoasidosizle kendisini belli eder. Hastalar anemi belirtisi de
gösterebilir. Sıklıkla akut ve ölümcül
olup bazı bireylerde kronik olarak devam eder.
Görülme Sıklığı
Türkiye’de sıklığı tam
olarak bilinmemektedir. Akraba evliliği olan hastalarda olma ihtimali
yüksektir. Bugüne kadar 450 den fazla vaka teşhis edilmiştir.
Hastalıkla İlgili Genler
MMUT , MMAA , MMAB , MCEE ve MMADHC bu
beş genin bir tanesinde anne ve babada taşıyıcı olması durumunda tanı
konulabilir.
Tedavi / Teşhis
Kritik hasta bireyler, hacim durumu ve asit-baz dengesini geri yükleyerek stabilize edilir; protein alımının azaltılması veya ortadan kaldırılması; katabolizmayı durdurmak için yüksek glikoz içeren sıvılar ve insülin yoluyla artan kalorilerin sağlanması; ve serum elektrolitleri ve amonyak, venöz veya arteriyel kan gazları ve idrar çıkışının izlenmesi. Yönetim, propiojenik amino asit öncülerinde düşük kalorili bir diyet içerir; hidroksokobalamin kas içi enjeksiyonları; karnitin takviyesi; bağırsak florasından propionat üretimini azaltmak için neomisin veya metronidazol gibi antibiyotikler; gerektiği gibi gastrostomi tüpünün yerleştirilmesi; ve enfeksiyonların agresif tedavisi. Sınırlı sayıda hastada kullanılan diğer terapiler arasında akut hiperammonemik atakların tedavisi için N- karbamilgluktamat; karaciğer, böbrek veya kombine karaciğer ve böbrek nakli; ve optik sinir atrofisinin tedavisi için antioksidanlar gibi yöntemler kullanılabilir.
Hastalıkta Kaçınılması Gereken
Oruç ve artan diyet proteinlerinden uzak durulması tavsiye edilir.
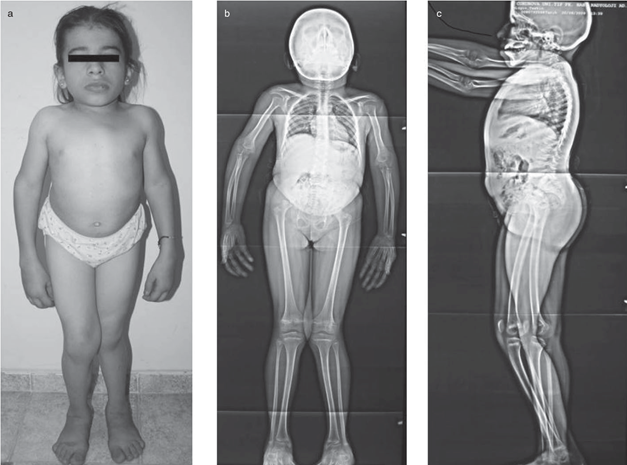
Turner sendromu, kızlar arasında en çok görülen kromozomal anomalilerden biridir. Nedeni, dişilerde 2 adet olması gereken X kromozomlarından birinin yokluğu ya da anormal olmasıdır. En tipik belirtileri, geniş ya da yele (perdeli) boyun (ekstra deri kıvrımları), bebeklerde şişmiş el ve ayaklar (lenfödem) , düz ve dışa dönük tırnaklar, iskelet anormallikleri veya böbrek problemleri vardır. Turner sendromlu bireylerin üçte biri ila yarısı, kalpten çıkan büyük arterin daralması (aort koarktasyonu) veya aortu kalbe (aort kapak) bağlayan kapak anormallikleri gibi bir kalp kusuru ile doğar. Bu kalp kusurları ile ilişkili komplikasyonlar hayatı tehdit edici olabilir. Turner sendromunun en yaygın özelliği, yaklaşık 5 yaşlarında ortaya çıkan kısa boydur. Erken yumurtalık fonksiyon kaybı (yumurtalık hipofonksiyonu veya erken yumurtalık yetmezliği) de çok yaygındır. Yumurtalıklar ilk başta normal olarak gelişir, ancak yumurta hücreleri (oositler) genellikle erken ölür ve çoğu yumurtalık dokusu doğumdan önce dejenere olur. Turner sendromlu çoğu kız ve kadın normal zekaya sahiptir. Bu özellikler etkilenen bireyler arasında farklılık gösterse de, gelişimsel gecikmeler, sözsüz öğrenme güçlükleri ve davranış problemleri mümkündür.
TS ilk olarak 1938’de Amerika Birleşik
Devletleri’nde Dr. Henry Turner tarafından tanımlanmıştır.
Genetik Değişiklikler /Etken Faktörler
Turner sendromu, iki cinsiyet
kromozomundan biri olan X kromozomu ile ilişkilidir. İnsanlar tipik olarak her
hücrede iki cinsiyet kromozomuna sahiptir: dişilerde iki X kromozomu
bulunurken, erkeklerde bir X kromozomu ve bir Y kromozomu bulunur. Turner
Sendromu, bir dişi hücrelerinde normal bir X kromozomu bulunduğunda ve diğer
cinsiyet kromozomu eksik olduğunda veya yapısal olarak değiştiğinde ortaya
çıkar. Eksik genetik materyal doğumdan önce ve sonra gelişimi etkiler.
Turner sendromlu bireylerin yaklaşık
yarısında monozomi X vardır, bu da bireyin vücudundaki her hücrenin normal iki
cinsiyet kromozomu yerine X kromozomunun yalnızca bir kopyasına sahip olduğu
anlamına gelir.
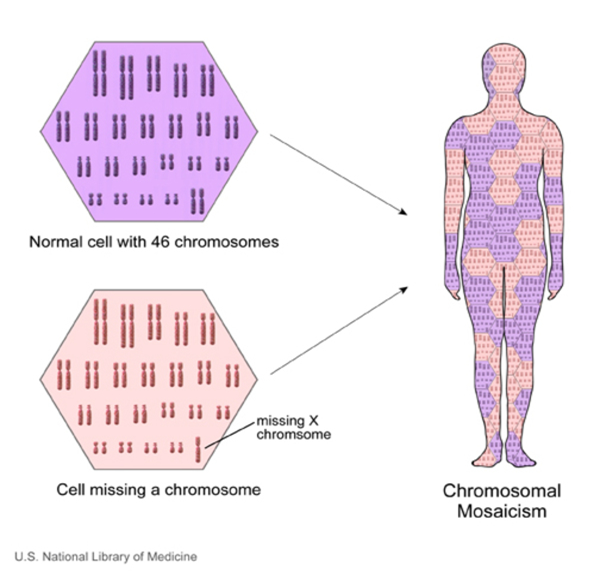
Turner sendromlu bazı kadınlar,
hücrelerinin sadece bazılarında mozaikçilik olarak bilinen kromozomal bir
değişikliğe sahiptir. X kromozomu mozaikçiliğinin neden olduğu Turner sendromlu
kadınların mozaik Turner sendromuna sahip olduğu söylenir.
Turner sendromunun
semptomları ve şiddeti bir kişiden diğerine oldukça değişebilir. Bozukluğun
birçok özelliği spesifik değildir ve diğerleri zamanla yavaşça gelişebilir veya
daha az belirgin olabilir. Etkilenen bireylerin aşağıda tartışılan semptomların
hepsine sahip olmayabileceğini belirtmek önemlidir. Etkilenen bireyler, özel
vakaları, ilişkili semptomları ve genel prognozu hakkında doktorları ve tıbbi
ekipleriyle konuşmalıdır.
Turner sendromlu hemen hemen
tüm kadınlar büyüme başarısızlığı gösterir ve ortalamadan daha kısa bir son
boyuta (kısa boy) ulaşır. Çocuklar başlangıçta, genellikle yaşamın ilk birkaç
yılında normal büyüme gösterebilir. Bununla birlikte, çoğu durumda, büyüme
oranı nihayetinde normalden daha yavaş olur ve etkilenen çocuklar normal büyüme
atakları yaşamazlar (örneğin ergenlik döneminde büyüme fışkırması olmaz).
Turner sendromunun bir diğer
yaygın özelliği, yumurtalıkların düzgün gelişememesidir (gonadal disgenez).
Gonadal disgenez, çocukluk döneminde erken yumurtalık fonksiyonunun kaybına
neden olabilir (erken yumurtalık yetmezliği). Normal olarak, yumurtalıklar
ergenlikte seks hormonları (örn. Östrojen ve progesteron) üretir. Bu hormonlar
ergenliğin başlaması ve ikincil cinsel özelliklerin doğru gelişimi için
gereklidir. Etkilenen kadınların çoğu, göğüsleri ve normal kadın vücudu
hatlarını geliştirmek, uygun kemik büyümesine girmek ve adet görmeye başlamak
için hormon replasman tedavisine ihtiyaç duyacaktır. Bazı durumlarda, etkilenen
bireyler meme gelişimine girebilir ve terapi olmadan menstruasyona başlayabilir
(spontan pubertal gelişim), ancak çoğu cinsel gelişimini durduracak ve gençlik
yıllarında daha sonraki bir noktada adet görmeyi durduracaktır.
Turner sendromlu dişiler,
perdeli bir görünüme sahip kısa bir boyun, başın arkasındaki düşük bir saç çizgisi,
düşük ayarlı kulaklar ve yukarı doğru çevrilmiş dar tırnaklar ve ayak
tırnakları gibi çeşitli farklı fiziksel özellikler geliştirebilir. Bazen
“kalkan sandığı” olarak adlandırılan geniş aralıklı meme uçlarına sahip geniş
bir göğüs oluşabilir. Bazı bireylerin şişmiş, kabarık elleri ve ayakları
olabilir. Bu belirtiler, lenfatik sistemi etkileyen bir durum olan lenfödem
nedeniyle ortaya çıkabilir. Lenfatik sistem, vücutta belirli protein açısından
zengin sıvı (lenf) ve kan hücrelerini filtreleyen ve dağıtan damarlar, kanallar
ve düğümlerin dolaşım ağıdır. Lenfödem, vücudun etkilenen bölgelerindeki sıvı
birikimine (ödem) bağlı şişme ile karakterizedir.
Ek fiziksel bulgular
arasında bir çene (retrognati), çapraz gözler (şaşılık), tembel gözler
(ambliyopi), sarkık göz kapakları ve ağızda darlık, yüksek kemerli bir çatı
(damak) sayılabilir. Bazı bireylerde ellerin kısa kemikleri, özellikle dördüncü
metakarplar, dirseklerde ortaya çıkan kollar ve düz ayaklar (pes planus) dahil
iskelet malformasyonları olabilir. Olguların yaklaşık% 10’unda, omurganın
anormal yanal eğriliği (skolyoz) de ortaya çıkabilir.
Bazı Turner sendromu
vakalarıyla ilişkili kalp kusurları, akciğerlerin arterlerinin yüksek tansiyonu
(pulmoner hipertansiyon) veya içte bir yırtık olan aort diseksiyonu da dahil
olmak üzere ciddi, hayatı tehdit eden komplikasyon riskini artırabilir.
Genetik Görülme Sıklığı
Bu durum dünya çapında 2.500 yeni doğan
kızdan yaklaşık 1’inde görülür, ancak terimde hayatta kalmayan hamileliklerde
(düşükler ve ölü doğumlar) çok daha yaygındır. Bozukluğun sıklığını etkileyen
bilinen herhangi bir ırksal veya etnik faktör yoktur. Bazı durumlarda, bozukluk
doğumdan önce veya doğumdan kısa bir süre sonra teşhis edilir. Bununla
birlikte, hafif vakalar yaşamın ilerleyen dönemlerine kadar ve hatta
yetişkinlik döneminde bile teşhis edilmeden kalabilir.
Kalıtım Paterni / Deseni
Turner sendromu vakalarının
çoğu kalıtsal değildir. Bu durum monozomi X’ten kaynaklandığında, kromozomal
anormallik, etkilenen kişinin ebeveyninde üreme hücrelerinin (yumurta ve sperm)
oluşumu sırasında rastgele bir olay olarak ortaya çıkar. Ayrılma adı verilen
hücre bölünmesindeki bir hata, anormal sayıda kromozomlu üreme hücrelerine
neden olabilir. Örneğin, bir yumurta veya sperm hücresi, ayrılmanın bir sonucu
olarak bir cinsiyet kromozomunu kaybedebilir. Bu atipik üreme hücrelerinden
biri çocuğun genetik yapısına katkıda bulunursa, çocuk her hücrede tek bir X
kromozomuna sahip olacak ve diğer cinsiyet kromozomunu kaçırmayacaktır.
Mozaik Turner sendromu da
kalıtsal değildir. Etkilenen bir kişide, erken fetal gelişimde hücre bölünmesi
sırasında rastgele bir olay olarak ortaya çıkar. Sonuç olarak, etkilenen bir
kişinin hücrelerinin bazıları normal iki cinsiyet kromozomuna sahiptir ve diğer
hücrelerde X kromozomunun yalnızca bir kopyası bulunur. X kromozom mozaikliği
olan kadınlarda diğer cinsiyet kromozom anormallikleri de mümkündür.
Nadiren, X kromozomunun
kısmen silinmesinin neden olduğu Turner sendromu bir nesilden diğerine
geçebilir.
Aşağıdaki bozuklukların
belirtileri Turner sendromunun semptomlarına benzer olabilir. Karşılaştırmalar
ayırıcı tanı için yararlı olabilir.
Noonan sendromu, doğumda tipik olarak görülen (konjenital)
yaygın bir genetik bozukluktur. Bozukluk, aralık ve şiddet açısından büyük
ölçüde değişen geniş bir semptom yelpazesi ve fiziksel özellik ile
karakterizedir. Etkilenen birçok kişide, ilişkili anormallikler ayırt edici bir
yüz görünümü içerir; geniş veya perdeli bir boyun; düşük bir posterior saç
çizgisi; tipik bir göğüs deformitesi ve kısa boy. Baş ve yüz (kraniyofasiyal)
bölgesinin karakteristik anormallikleri, genişçe ayarlanmış gözleri (oküler
hipertelorizm); gözlerin iç köşelerini kaplayabilen cilt kıvrımları (efsanevi
kıvrımlar); üst göz kapaklarının sarkması (pitoz); küçük bir çene (mikrognati);
depresif bir burun kökü; geniş tabanlı kısa bir burun; ve düşük yerleşimli,
arkaya döndürülmüş kulaklar (pinnae). Göğüs kemiği (sternum) anormallikleri,
omurganın eğriliği (kifoz ve / veya skolyoz) ve dirseklerin dışa doğru sapması
(cubitus valgus) gibi belirgin iskelet malformasyonları da tipik olarak
mevcuttur. Noonan sendromlu birçok bebekte ayrıca, kalbin sağ alt odasından
akciğerlere (pulmoner kapak darlığı) uygun kan akışının engellenmesi gibi kalp
(kardiyak) kusurları vardır. Ek anormallikler, belirli kan ve lenf damarlarının
malformasyonlarını, kan pıhtılaşmasını ve trombosit eksikliklerini, öğrenme
güçlüklerini veya hafif zihinsel özürlülüğü, testislerin etkilenen erkeklerde
yaşamın ilk yılına kadar inmesini (ve kriptorşidizmi) içerebilir ve / veya
diğer semptom ve bulgular. Noonan sendromu, rasopati yolunu oluşturan çoklu
tekli genlerdeki anormalliklerin (mutasyonlar) neden olduğu otozomal dominant
bir genetik bozukluktur. Noonan sendromu ile ilişkili bazı semptomlar, Turner
sendromu olanlara yüzeysel olarak benzeyebilir (kısa boy, perdeli boyun vb.
Gibi her iki bozuklukla ilişkili olabilecek bazı bulgular nedeniyle). Sonuç
olarak, geçmişte, Noonan sendromu “erkek Turner sendromu”, “kadın yalancı
Turner sendromu” veya “normal kromozomlu karyotipli Turner fenotipi” olarak
anılmıştır. Bununla birlikte, iki bozukluk arasında birçok önemli fark vardır.
Noonan sendromu hem erkekleri hem de kadınları etkiler ve normal bir kromozomal
makyaj (karyotip) vardır. X kromozomunu etkileyen anormallikler ile karakterize
edilen Turner sendromundan sadece kadınlar etkilenir.
Teşhis
Ayrıntılı bir hasta öyküsü,
kapsamlı bir klinik değerlendirme ve çeşitli özel testler. Büyüme eksikliği
veya nedeni bilinmeyen kısa boylu kızlarda Turner sendromundan
şüphelenilmelidir.
Turner sendromu tanısı
genellikle karyotip belirlenerek elde edilen kromozomal analizle doğrulanır.
Karyotipleme, kromozomların sayısını ve yapısını değerlendiren bir laboratuvar
testidir.
Bazı durumlarda, fetal
ultrasonda Turner sendromu ile ilişkili bazı fiziksel bulgular görülebilir.
Örneğin, gelişmekte olan bir fetüsün boynuna yakın lenf sıvısı birikimi bazen
rutin bir fetal ultrasonda görülebilir. Kesin test CVS veya amniyosentez ile
yapılabilir. CVS, gebeliğin 10-12. haftasında yapılır ve doku örneklerinin
plasentanın bir kısmından çıkarılmasını içerirken, amniyosentez 16-18. gebelik
haftasında yapılır ve fetüsün etrafında küçük bir sıvı örneği almayı içerir.
Etkilenen bireyleri
karaciğer, böbrek veya kalp anormallikleri gibi Turner sendromuyla ilişkili
potansiyel semptomların varlığı açısından değerlendirmek için manyetik rezonans
görüntüleme (MRI) gibi spesifik görüntüleme teknikleri uygulanabilir.
Tiroid ve karaciğer
fonksiyonu, kemik yaşı ve büyüme hakkında ek değerlendirme yapılmalıdır.
Hipertansiyon taraması da yapılmalıdır. Doğumda teşhis edilen bebeklere işitme
muayenesi de dahil olmak üzere tam bir kulak, burun ve boğaz muayenesi
yapılmalıdır. Çocuklar, özellikle tekrarlayan orta kulak iltihabı yaşayanlar ve
yetişkinler periyodik işitme değerlendirmesi gerektirir.
Tedavi
Turner sendromunun tedavisi,
her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, bir
uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, çocuk
uzmanları, cerrahlar, kardiyologlar, endokrinologlar, konuşma patologları,
kulak burun boğaz uzmanları, göz doktorları, psikologlar ve diğer sağlık
uzmanlarının sistematik ve kapsamlı bir şekilde çocuğun tedavisini etkilemesi
gerekebilir. Etkilenen bireyler ve aileleri için genetik danışmanlık önerilir.
Belirli ilaç rejimlerinin ve
/ veya diğer tedavilerin kullanımına ilişkin kararlar, hekim ve sağlık ekibinin
diğer üyeleri tarafından, sendromun özelliklerine dayanarak hastayla dikkatli
konsültasyonda verilmelidir; olası yan etkiler ve uzun vadeli etkiler de dahil
olmak üzere potansiyel faydalar ve riskler hakkında kapsamlı bir tartışma
yapılıp; hasta tercihi ve diğer uygun faktörler değerlendirilmelidir.
Turner sendromu için bir
tedavi yoktur, ancak fiziksel gelişimi artıracak tedaviler geliştirilmiştir.
Uygun tıbbi bakım ile Turner sendromlu kadınlar tam, üretken yaşamlar
sürdürebilmelidir. Etkilenen bireyler için birincil tedaviler büyüme hormonu tedavisi
ve östrojen tedavisidir. Çocuklardaki büyüme hormonu tedavisi Çocuk
endokrinoloji hekimlerince uygun görülen sürede planlanır.
Hormon replasman tedavisi genellikle 12-14
yaşlarında başlar. Çoğu ortalama kız ergenliğe girecek. Ergenliğin
başlatılmasının zamanlaması, büyüme hormonu replasmanındaki büyüme ilerlemesini
de dikkate alır. Bu özellikleri korumak için replasman tedavisine devam
edilmelidir ve çoğu kadın menopoza kadar östrojen ve progesteron tedavisine
ihtiyaç duyacaktır.
Ek tedavi semptomatik ve
destekleyicidir. Örneğin, tiroid hormonu replasman tedavisi tiroid hastalığı
olan bireyleri tedavi etmek için kullanılabilir. İşitme kaybının işitme
cihazlarıyla düzeltilmesi, öğrenme ve sosyal etkileşime yardımcı olabilecek bir
diğer önemli müdahaledir.
Turner sendromlu çocukların
potansiyellerine ulaşmalarını sağlamak için erken müdahale önemlidir.
Tıp uzmanlarının ve iyi bir
sosyal destek sisteminin yardımıyla, TS’li bir kadın tatmin edici, sağlıklı bir
yaşam sürmeyi bekleyebilir.
Schimke immüno-ossöz displazisi , kısa boy, böbrek hastalığı ve zayıf bir bağışıklık sistemi ile karakterize bir durumdur. Bu durumu olan insanlarda, kısa boy düzleşmiş omurga kemiklerinden ( vertebralar) kaynaklanır.), Görsel 1kısaltılmış boyun ve gövde ile sonuçlanır. Yetişkin yüksekliği tipik olarak 3 ila 5 feet arasındadır. Böbrek (böbrek) hastalığı sıklıkla hayatı tehdit eden böbrek yetmezliğine ve son dönem böbrek hastalığına (ESRD) yol açar. Etkilenen bireyler ayrıca T hücreleri adı verilen bazı bağışıklık sistemi hücrelerinin yetersizliğine de sahiptir.. T hücreleri yabancı maddeleri tanımlar ve vücudu enfeksiyonlara karşı korur. T hücrelerinin yetersizliği, bir kişinin hastalığa karşı daha duyarlı olmasına neden olur.
Bu rahatsızlığı olan
kişilerde sık görülen diğer özellikler arasında alt sırtın abartılı bir
eğriliği vardır ( lordoz); tipik olarak
göğüste ve sırtta koyulaşmış cilt lekeleri (hiperpigmentasyon); ve burnun
yuvarlatılmış ucu olan geniş bir burun köprüsü.
Schimke immüno-ossöz displazinin daha az görülen belirtileri ve semptomları ,
atardamarların astarında ( ateroskleroz) yağ
birikintileri ve skar benzeri bir doku birikimini içerir.) beyine kan akışını
azalttı (beyin iskemisi), migren benzeri baş ağrıları, yetersiz tiroit bezi (hipotiroidizm), azalmış beyaz kan hücreleri (lenfopeni), az
gelişmiş kalça kemikleri (hipoplastik pelvis), anormal derecede küçük baş
büyüklüğü ( mikrosefali)),
erkeklerde sperm eksikliği (azospermi) ve kadınlarda düzensiz adet kanaması.
Şiddetli vakalarda, doğumda Schimke immüno-ossöz displazinin birçok
belirtisi mevcut olabilir. Bu hastalığın hafif vakaları olan kişiler
geç çocukluğa kadar belirti veya semptom geliştirmeyebilir.
Schimke immuno-ossöz displazili hastaların yaklaşık yarısında SMARCAL1 geninde mutasyon
tanımlanmamıştır. Bu gibi durumlarda, hastalığın nedeni bilinmemektedir.
Bazı insanlar erken çocukluk döneminde şiddetli bir form
geliştirirken, diğerleri çocuklukta veya sonrasında hafif bir form
geliştirir. Kısa boy, omurganın anormal gelişimini ve uzun kemiklerin
uçlarını içeren spondiloepifizeal displaziden kaynaklanır . Neredeyse SIOD’lu
tüm kişilerin böbrek hastalığı vardır, bu da böbrek hastalığına son aşamada ilerler.
Schimke immünoosöz displazisi (SIOD) şiddetine göre değişmektedir. Erken
başlangıç formuna sahip olanlar genellikle ciddi semptomlara ve yaklaşık 9
yıllık ortalama ömre sahiptir. Ölüm nedenleri inme, ciddi fırsatçı
enfeksiyon, kemik iliği yetmezliği, böbrek yetmezliği komplikasyonları,
konjestif kalp yetmezliği veya akciğer hastalığını içerebilir. Daha hafif
belirtileri olanlarda yetişkinlik çağında hayatta kalırlar, böbrek hastalığı
iyi yönetilerek. Bununla birlikte, başlangıç şiddeti ve yaşının mutlaka
yaşam beklentisini kesin göstermeyeceğini not etmek önemlidir, çünkü şiddetli,
erken başlangıçlı SIOD’lu bazı insanlar 20’li ve 30’lu yaşlarında hayatta
kalmıştır.
Genetik Değişiklikler /Etken Faktörler
SMARCAL1 genindeki mutasyonlar, Schimke immüno-osöz displazisi riskini
arttırır. SMARCAL1 gen olan spesifik fonksiyonu bilinmeyen bir protein üretmek için
talimatlar sağlar. SMARCAL1
proteini kromatine bağlanabilir
DNA’yı kromozomlara paketleyen DNA ve protein kompleksidir. Benzer
proteinlerin fonksiyonuna dayanarak, SMARCAL1’in, kromatin remodeling olarak
bilinen bir işlemle diğer genlerin aktivitesini (ekspresyonunu) etkilediği
düşünülmektedir. Kromatinin yapısı, DNA’nın ne kadar sıkı bir şekilde
paketlendiğini değiştirmek için değiştirilebilir (yeniden
yapılandırılabilir). Kromatin remodeling, gelişim sırasında regresyonun
düzenlenmesinin bir yoludur. DNA sıkıca paketlendiğinde, gen ekspresyonu,
DNA’nın gevşek bir şekilde paketlendiğinden daha düşüktür.
SMARCAL1 genindeki mutasyonların, protein
aktivitesini, protein stabilitesini veya proteinin kromatine bağlanma
yeteneğini etkileyerek hastalığa yol açtığı düşünülmektedir. SMARCAL1 genindeki mutasyonların kromatin
remodelingine ve diğer genlerin ekspresyonuna müdahale edip etmediği açık değildir.
Schimke immüno-disöz displazisi ile ilişkili mutasyonlar, SMARCAL1 proteininin olağan
fonksiyonlarını bozar veya herhangi bir fonksiyonel proteinin üretimini
önler. Fonksiyonel bir protein eksikliğine neden olan mutasyonlara sahip
olan insanlar, aktif fakat hatalı çalışan bir proteine yol açan mutasyonlara
sahip olanlardan daha şiddetli bir şekilde bu hastalığa sahip olma
eğilimindedir. Bununla birlikte, SMARCAL1 gen
mutasyonlarına sahip kişilerin Schimke immüno-kemikli displazisi geliştirmesi
için , şu anda bilinmeyen diğer genetik veya çevresel faktörlerin de
bulunması gerekir.
Belirti ve Semptomlar
Schimke
immünoosöz displazisi (SIOD) vücudun birçok bölümünü etkiler. Yavaş büyüme
genellikle SIOD’un ilk işaretidir. İnsanlar genellikle kısa bir boyun ve
gövde geliştirir (orantısız şekilde).kısa
boy) spondiloepifizeal displazi nedeniyle . Kemik
anormallikleri tipik olarak omurga veya kalçalarda gelişir. SIOD’lu birçok
kişi sonunda kalça
protezi ameliyatına ihtiyaç
duyar .Böbrek hastalığı tanı anında var olur veya birkaç
yıl içinde gelişir ve sonuçta böbrek yetmezliğine ilerler . Neredeyse
tüm SIOD’lu kişilerin kanı varhücreEn sık T-hücrelerinin
eksikliği. Bu, hayati tehlike oluşturabilecek enfeksiyon riskinin
artmasına neden olur.
Schimke immuno-osseöz displazisi çok nadir görülen bir
durumdur. Kuzey Amerika’da yaygınlığın 1 milyondan 3 milyona kadar bir
insan olduğu tahmin edilmektedir.
Kalıtım Paterni\Deseni
SMARCAL1 genindeki mutasyonlar, otozomal resesif paternde kalıtsaldır bu, Schimke
immüno-ossöz displazisi riskinin artmasının,
her hücrede SMARCAL1 geninin
her iki kopyasındaki mutasyonlardan kaynaklandığı anlamına
gelir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri
mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun
belirtilerini ve semptomlarını göstermezler.
Kavramsal olarak, etkilenen
bir bireyin her
birinin % 25’i etkilenme şansı, %50’sinin asemptomatik bir taşıyıcı olma olasılığı ve % 25’inin de
etkilenmeme ve taşıyıcı olmama olasılığı vardır. Ailede patojenik
varyantların her ikisi de biliniyorsa taşıyıcı testi ve doğum öncesi testi
mümkündür.
Teşhis Yöntemleri ve Tedavileri
Teşhis:
Aşağıdaki temel
özelliklere sahip kişilerde SIOD tanısından şüphelenilir:
Tanı klinik muayeneden
sonra konulabilir. Genetik test tespit
etmek mutasyonlariçinde SMARCAL1 genteşhisi
onaylayabilir.
Kıkırdak kılı hipoplazisi (bu terime bakınız) ana
ayırıcı tanıdır.
Tedavi:
Schimke
immunoosseöz displazinin (SIOD) tedavisi her bireyin şiddetine ve bireysel
semptomlarına bağlıdır. Kalça, böbreklerin düzenli olarak izlenmesi,bağışıklık sistemive kan tavsiye edilir.
SIOD için gerekli olabilecek tedavi örnekleri
şunları içerir:
Böbrek diyaliz veya
nakli
Kalça protezi
Nötropeninin
granülosit ile tedavisi koloni uyarıcı faktör veya
granülosit-makrofaj koloni uyarıcı faktör
Kemik iliği
nakli immün yetmezlik için (transplantasyon sonrası birkaç ölüm
bildirilmiş olmasına rağmen)
Otoimmün semptomları
olanlar için bağışıklık sistemini baskılayan ilaçlar
Şiddetli
papilloma tedavisinde Imiquimod ve cidofovir (bir
antiviral)virüs cilt enfeksiyonları
Geçici iskemik
atakları (“mini vuruş”) veya felç tedavisinde kan akışını iyileştiren
veya kanın pıhtılaşmasını azaltan ajanlar
Hipotiroidizmin standart
tedavisi
Standart
tedavi skolyoz
Hastalıkla İlişkili Genler
Schimke immüno-ossöz displazisi (SIOD),
spondiloepifizeal displazi ve orantısız kısa boy, yüz dismorfizmi, T hücreli
immün yetmezlik ve nefrotik sendromlu glomerülonefrit ile karakterize multisistemik
bir hastalıktır.
SIOD , SMATCAL1 genindeki (2q35), kromatin remodeling proteini hHARP’yi
(ayrıca kromatin alt-ailesi A-benzeri protein 1’in SWI / SNF ile ilgili
matris-ilişkili aktin bağımlı regülatörü olarak da bilinir)
kodlayan mutasyonlardan kaynaklanır .
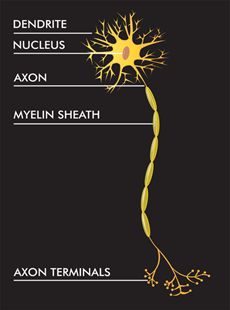
Pelizaeus-merzbacher hastalığı daha çok erkekleri etkileyen beyin ve omuriliğini (spinal cord- merkezi sinir sistemi) kapsayan kalıtsal bir durumdur. Bu hastalık lökodistrofi denilen genetik hastalık gruplarından birisidir. Lökodistrofiler, miyelin adı verilen yağlı bir madde ile kaplanmış sinir liflerinden oluşan sinir sistemi beyaz maddesindeki anormallikleri içeren durumlardır. Miyelin sinir liflerini korur ve sinir impulslarına hızlı bir geçiş olanağı sunar. Özellikle pelizaeus-merzbacher hastalığında hipomiyelinasyon görülür ve hipomiyelinasyonda sinir sisteminde miyelin üretimi azalır. Sonuç olarak da sinir sistemi fonksiyonlarında düşüş meydana gelir.
Pelizaeus-merzbacher
hastalığı klasik ve konnatal (doğuştan) olmak üzere iki şekilde
sınıflandırılır. Bu iki çeşidin ciddiyet derecesi farklılık gösterse de
özellikleri benzerdir.
Klasik
pelizaeus-merzbacher daha sık karşılaşılan bir tiptir. Yaşamın ilk yılında bu
hastalık grubundaki bireylerde zayıf kas tonusu (hipotonia); istemsiz oluşan
göz hareketleri (nystagmus) ve oturmak, cisimleri kavramak gibi motor
becerilerde gecikme görülür. Bazı bireyler yardım alarak yürüyebilir. Tüm bu
nörolojik problemlere rağmen zihinsel ve motor beceriler gelişmeye devam eder
ancak büyüme çağıyla beraber bu gelişme durur ve yavaş yavaş kaybolmaya başlar.
Durum ilerledikçe nystagmus ortadan kaybolabilir ancak kas katılığı
(spastisite), hareket ve denge problemleri (ataksia), baş ve boyunda titreme
(titübasyon), istemsiz kas kasılmaları (distonia) ve ani hareketler (koreiform)
gibi problemler devam eder.
Konnatal PMD ise bu
iki tipten en ağır olan formdur. Semptomlar çocuklukta başlar ve beslenme
problemleri, kilo kaybı ve yavaş büyüme, solunum yolu tıkanıklığı nedeniyle zor
nefes alıp verme, nystagmus, ilerleyen konuşma bozuklukları (disartria), ciddi
ataksia, hipotonia ve nöbetler ile devam eder. Durum kötüleştikçe hasta çocukta
hareket etmeyi sınırlayan eklem deformitesine neden olan spastisite gelişir. Bu
tipteki hastalar asla yürüyemez ve kollarını istemli olarak hareket ettiremezler.
Konuşma problemi yaşarlar ancak karşılarındakini anlayabilirler.
Görülme Sıklığı
Klasik ve konnatal PMD formları erkek bireylerde
kadınlara oranla daha yüksek oranda görülür. Amerika Birleşik Devletleri’nde
pelizeaus-merzbacher hastalığının görülme sıklığı 200.000-500.000 erkek bireyde
birdir.
Nedenleri
PLP1 genindeki mutasyonlar
pelizeaus-merzbacher hastalığına neden olmaktadır. PLP1 geni proteolipit
protein 1 üretimini yönetir ve DM20 proteininin modifiye (isoform) formudur.
Proteolipid protein 1 başlıca sinir sistemindeki sinirlerde; DM20 proteini ise
beyin ve omuriliği kaslara bağlayan sinirlerde (periferal sinir sistemi)
bulunur. Bu iki protein miyelinin büyük bir kısmını oluşturan hücre membranında
yer alırlar ve onu hücrelere bağlarlar. Pelizeaus-merzbacher hastalığına sebep
olan birçok mutasyon PLP1 genini duplikasyona uğratır ve bunun sonucunda
proteolipid protein 1 ve DM20 protein üretimi artar. Diğer mutasyonlar genellikle
yanlış katlanan protein üretilmesine neden olur. Fazla ya da anormal proteinler
hücre tarafından tutulur ve membran içerisinde seyahat edemez. Sonuç olarak
proteolipid1 ve DM20 proteinleri miyelin kılıfı oluşturamazlar. Fazla
proteinlerin birikimi sinir liflerinde şişme ve bozulmalara yol açar. Yine
başka mutasyonlar, proteolipid protein 1 ve DM20 protein üretimini önleyen ve
hücre zarı içinde bu proteinlerin eksikliğine neden olan ve bu şekilde oluşan
herhangi bir miyelinin, kararsız hale gelmesine ve hızlı bir şekilde
parçalanmasına neden olan PLP1 genini siler. Bütün bu PLP1 gen mutasyonları
hipomiyelinasyon, sinir lifi hasarı ve sinir sistemi fonksiyonunda zayıflamaya
neden olarak hastalığın belirti ve semptomlarını meydana getirir. Pelizaeus-Merzbacher
hastalığı olan kişilerin yüzde 5 ila 20’sinin PLP1 geninde tanımlanmış
mutasyonlara sahip olmadığı tahmin edilmektedir.
Kalıtım Paterni
Pelizaeus-merzbacher
X kromozomuyla kalıtılan bir hastalıktır. Yalnızca bir tane X kromozomuna sahip
erkeklerde mutant PLP1 geninin bir kopyası hastalığın ortaya çıkması için
yeterlidir. İki tane X kromozomu bulunan kadınlarda ise değişmiş genin bir
kopyası kas katılığı veya düşük zihinsel fonksiyonlar gibi daha az ciddiyette
semptomlara neden olabilir ya da hiçbir semptom ve belirti ortaya çıkmayabilir.
Hastalığın kalıtımı anneden gelen genlerle sağlanır. Mutasyon bazen
kalıtılırken bazen de çevresel faktörlerle kendi kendine oluşur.
Diğer İsimleri
Cockayne-Pelizaeus-Merzbacher
disease
HLD1
Hypomyelinating
leukodystrophy, 1
PMD
Sudanophilic
leukodystrophy
Belirti ve Semptomlar
PMD’nin belirtileri
kişiden kişiye farklılık gösterebilir. Başlangıçta, hasta çocuklar baş ve göz
hareketlerinin normal kontrolünde başarısız olur, özellikle istemsiz ve hızlı
baş ve göz (nystagmus) hareketleri görülür. Anormal yavaş gelişim de erken
belirtiler arasındadır. Hasta çocuk büyüdükçe kas titremeleri, zayıf kas tonusu
(hipotoni), istemli hareketlerin kontrolündeki zayıflık gibi diğer belirtiler de
ortaya çıkmaya başlar. Etkilenen bireylerde ayrıca bacaklarda yavaş, sert
hareketler ve kolların ve bacakların potansiyel kısmi felci (spastik
quadriparesis) ile sonuçlanan istemsiz kas spazmları (spastisite); belirli
eklemlerin anormal, kalıcı olarak sabitlenmesi (kontraktürler); göze giden
sinirlerin ilerleyen dejenerasyonu (optik atrofi); ve / veya konuşma zorluğu
(dizartri) gelişebilir. Bazı durumlarda, etkilenen çocuklar büyüdükçe nystagmus
kaybolabilir. Bazı çocuklarda genellikle zamanla artan şiddetli spastisiteye ek
olarak iskelet deformiteleri de gelişebilir
Transisyonel PMD ise
konnatal ve klasik PMD arasındaki geçiş formudur. Etkileri her ikisiyle de
benzerdir. Ancak klasik formdan daha hızlı ilerleme gösterirken konnatal
formdan daha yavaştır.
Teşhis
PMD tanısı, ayrıntılı
bir klinik değerlendirme, hasta geçmişi ve beyaz madde eksikliğini saptamak
için manyetik rezonans görüntüleme (MRG) gibi çeşitli özel testlere dayanarak
konulabilir. Beyincik (cerebellum) ve beyin kökündeki miyelinasyon eksikliği
gibi durumların erken tanısı PMD formlarının tedavisine yardımcı olabilir.
Moleküler genetik testleri PLP genini taramak için kullanılabilir.
Tedavi
PMD tedavisi için
standart bir tedavi yöntemi yoktur. Yalnızca nöbetleri kontrol altına almak
için ilaç tedavisi ya da hareket bozuklukları için fiziksel rehabilitasyon
yöntemleri uygulanabilir. Ancak
hastalığı kontrol altında tutabilmek için erken teşhis önemli rol oynamaktadır.





{kind=link}
{kind=link}
{kind=link}