Antisynthetase sendromu; kronik bir otoimmün durumudur. Vücudun kaslarını ve çeşitli kısımlarını etkiler. Birçok belirti ve semptomu kapsar. Kas iltihabı (miyozit), poliartrit (birçok eklem iltihabı), interstisyel akciğer iltihabı, ellerin kalınlaşması ve çatlaması ve Raynaud fenomenini kapsayana karakterize bir durumdur. Esase vakanın altında yatan neden bilinmemektedir ancak vücutta biyosentezi olan aminoasil-tRNA sentetazları adındaki enzimlere saldıran otoantikoların (normal antikorlara saldıran antikorlar) üretiminin, sendromun nedeni olarak düşünülen kanılardan birisidir. Bu otoantikorlar viral enfeksiyonla beraber ortaya çıkabilir veya viral enfeksiyondan sonra da ortaya çıkabilir. Hastaların genetik yatkınlığı da söz konusu olabilir. Tedavi, her insanda belirti ve semptomlara dayanmaktadır; kortikosteroidleri, immünosüpresif ilaçlar veya fizik tedaviyi barındırabilir.
Antisynthetase sendrom hastalarının uzun süre bakımı, kronik immünosüpresif tedavinin olumsuz etkilerine ve kompilasyonlarına maruz kalabilir. Pulmoner hipertansiyon, malignite ve azalmış sağkalımı gerektiren ilerleyici interstisyel akciğer hastalığını içerebilen hastalıkla ilişkili doku bozuklukları beraberinde getirir.
Akciğer transplantasyonu, pulmoner hipertansiyon, malignite ve azalmış sağkalımı gerektiren doku bozukluğu olan interstisyel akciğer hastalığının klinik özellikleri hakkında daha fazla tanı, araştırma, tanı, teşhis, tedavi ve daha fazla farkındalığın oluşumu Antisynthetase sendrom hastaları için umud vaad edilmektedir.
Antisynthetase Sendrom Semptomları:
Kas
iltihabı (miyozit)
Poliartrit
(bi kaç eklemde iltihap)
Ateş
Mekanik
eller
Raynaud
Fenomeni
İnterstisyel
akciğer hastalığı (spesfik olmayan akciğer iltihabı)
Başlıca
semptomlar bunlarla beraberinde olup zamanla değişme ihtimali de vardır.
Antisynthetase Sendromu Tanısı Nasıl Yapılır?
Kas enzimleri, örneğin kreatinin
kinaz (CK) ve aldolaz: bunlar genellikle yükselir
Kas antikorları
Elektromiyografi (EMG)
Etkilenen kasların manyetik
rezonans görüntüleme ( MRI )
Kas biyopsisi
Akciğer fonksiyon testleri
Göğsün yüksek
çözünürlüklü bilgisayarlı tomografi taraması ( BT )
Yutma güçlükleri ve aspirasyon
riskinin değerlendirilmesi
Akciğer biyopsisi
Antisintaztaz Sendromu Nasıl Tedavi Edilir?
Glukokortikosteroidler antisynthetase
sendromu için tedavinin temel dayanağıdır ve genellikle birkaç ay veya yıl
boyunca gereklidir. Prednizon başlangıçta 4-6 hafta boyunca yüksek
dozlarda (1 mg / kg / gün) hastalık kontrolünü sağlamak için verilir,
daha sonra remisyonu sürdürmek için en düşük etkili doza 9-12 ay
boyunca yavaşça azaltılır . Daha ciddi vakalarda, 3-5 gün
boyunca darbeli intravenöz (IV) metilprednizolon gerekebilir.
Steroid kaynaklı osteoporoza ve Pneumocystis jirovecii gibi bazı mantar enfeksiyonlarına karşı profilaktik tedavi önerilir . Tedaviye başlamadan önce aşılama ihtiyacı değerlendirilmelidir.
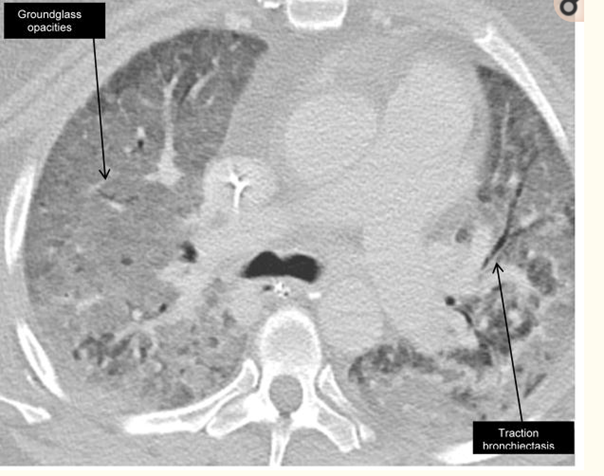
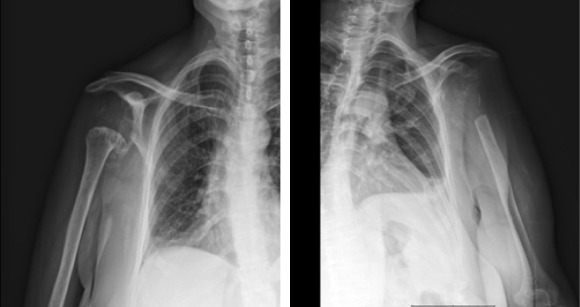
Şekil 1) Antisynthetase sendrom yüksek çözünürlüklü BT’si.
The Diagnosis and Treatment of Antisynthetase Syndrome Leah J. Witt, MD,1 James J. Curran, MD,2 and Mary E. Strek, MD1 PMCID: PMC500639 NIHMSID: NIHMS80052PMID: 27594777
Konjenital
alfa2 antiplazmin eksikliği,
küçük travma ve spontan kanama epizodlarını takiben uzun süreli kanama ve
ekimozların neden olduğu konjenital alfa2 antiplazmin eksikliğinin neden olduğu
nadir bir hemorajik bozukluktur (genellikle uzun kemiklerin diyafizi gibi
alışılmadık yerlerde). Konjenital alfa2 antiplazmin eksikliği otozomal
resesif bir şekilde kalıtsaldır.
Konjenital alfa2 antiplazmin
eksikliği, ilk olarak 1969 yılında Japonya’nın güneybatısındaki Okinawa
adasında yaşayan bir çocukta Masateru Kohakura tarafından
tanımlanmıştır. Çocuğun kesiklerden uzun süreli kanama, subkutan kanamalar
ve travmatik eklem kanaması gibi hemorajik diyatezi vardı. Daha sonra, tüm
soyağacı, ortak atalardan gelen aile üyeleri arasında ve hepsi adada yaşayan 3
akraba evliliği ile tanımlandı. alfa 2-antiplazmin, insan dolaşımda plazmin
doğal inhibitörüdür ve fibrinoliz düzenlenmesinde önemli bir rol
oynar. Plazmin-antiplazmin sistemi, fibrin polimerlerinin çözünür
fragmanlara çözülmesini düzenler.
Genetik Değişiklik/Etken Faktörler
Alfa 2-antiplazmin eksikliği konjenital veya edinsel olabilir. Konjenital alfa 2-antiplazmin eksikliği olan hastalar, plazmin inaktivasyonu ve hemostatik fibrin tıkacının erken lizizine bağlı olarak ciddi bir hemorajik bozukluk ile başvurabilir. Travma veya ameliyattan sonra bazen kanama gecikebilir.
Edinilmiş alfa 2-antiplazmin eksikliği karaciğer hastalığı (sentez azalması), yaygın damar içi pıhtılaşma (artan tüketim), nefrotik sendrom (idrar kaybı), amiloidoz veya trombolitik tedavi sırasında görülebilir.
Alfa-2-plazmin inhibitörü
eksikliğine PLI (SERPINF2; 613168 ) genindeki mutasyonun neden olduğu
vakalarda tespit edilmiştir.
Belirti ve Semptomlar
Homozigot alfa 2-antiplazmin eksikliği olan hastalar göbek kordonundan kanama ile çocuklukta erken ortaya çıkabilir ve ömür boyu kanama eğilimi gösterir.
Kanama belirtileri konjenital
hemofili olan bir hastaya benzer ve aşağıdakileri içerebilir:
Minör travma sonrası gecikmiş kanama başlangıcı
Mukozal kanama dahil olmak üzere kesik ve
yaralardan uzun süreli kanama
Kas kanaması dahil artan morarma ve hematomlar
Travma sonrası spontan eklem kanaması yerine
eklemlere kanama
Ameliyat sonrası aşırı kanama (daha hafif
vakalarda ipucu olabilir)
Aşırı morarma
Steroid olmayan antienflamatuar ilaçların
(NSAID’ler) kullanımından sonra artan kanama
Bademcik ameliyatı, adenoidektomi ve diş çekimi
sonrası uzun süreli kanama bildirilmiştir.
Etkilenen bireylerde spontan
hemotoraks, hematüri, burun kanaması ve kas hematomları da
tanımlanmıştır. Uzun kemiklerin diyafizinde olağandışı intramedüller
kanaması olan birçok hasta, kendiliğinden veya travma ile ortaya
çıkmıştır.
Heterozigot alfa 2-antiplazmin eksikliği daha hafif kanama epizodları ile ilişkili olabilir ve çoğu hasta asemptomatik olabilir. Diş veya cerrahi prosedürlerden sonra uzun süreli kanama meydana gelebilir. Alfa 2-antiplazmin seviyelerinin düşmesinin bir sonucu olarak, ilerleyen yaşla birlikte kanama belirtileri ortaya çıkabilir.
Genetik Görülme Sıklığı
Alfa 2-antiplazmin eksikliği
dünya çapında tarif edilen sadece bir avuç vaka ile çok nadir görülen bir
hastalıktır. Sonuç olarak, bu bozukluğun yaygınlığı ve etnik tercihleri
bilinmemektedir. Yüksek verimli genomik testlerin daha yaygın kullanımı
ile, bu durumun sıklığı hakkında daha fazla bilgi elde edilebilir.
Kalıtım Paterni/Deseni
Alfa 2-antiplazmin konjenital
eksikliği, otozomal resesif bir kalıtım paternini takip eder. Homozigot
eksikliği olan hastaların ailelerinde akraba evliliği öyküsü vardır.
Birkaç alellik varyant
tanımlanmıştır.
Teşhis Yöntemleri ve Tedavileri
Teşhis
Alfa 2-antiplazmin eksikliği
için spesifik bir tahlil, bir hastanın olağan tarama tahlilleri ile
tanımlanamayan bir kanama diyatezi olduğunda yapılır. Hem alfa
2-antiplazmin antijeni hem de aktivite seviyeleri ölçülebilir.
İki tür alfa 2-antiplazmin
eksikliği vardır:
Tip 1 – Azalan antijen ve aktivite seviyeleri
Tip 2 – Normal antijen seviyesi ile düşük
aktivite seviyesi
Homozigot hastalar genellikle
saptanamayan seviyelere sahipken heterozigot hastalar normal seviyelerin% 40 -%
60’ına sahiptir. Normal plazma konsantrasyonu 0.7 mg / L’dir.
Tedavi
Alfa 2-antiplazmin ile
ilişkili kanama genellikle aminokaproik asit veya traneksamik asit gibi
antifibrinolitik ajanlarla etkili bir şekilde tedavi edilir. Bu ajanlar
herhangi bir cerrahi veya diş prosedüründen önce kanamaya yanıt olarak veya
profilaksi olarak kullanılabilir. Antifibrinolitik ajanlar esas olarak
plazminojenin fibrine bağlanmasını önleyerek, endojen fibrinolizi inhibe ederek
ve hemostatik tıkacı stabilize ederek etki gösterir. Oral veya intravenöz
olarak verilebilirler ve genellikle iyi tolere edilirler.
Strongyloidiasis , yuvarlak kurt Strongyloides
stercoralis’in (S. stercoralis) neden olduğu parazitik bir hastalıktır.
İnsanlar, solucanlarla kontamine olmuş toprakla temas ettiklerinde enfeksiyonu
yakalarlar. Sıklıkla semptom
görülmezken, karın ağrısı, öksürük, ishal, döküntü, nedensiz kilo kaybı ve
kusma görülebilir. Enfeksiyon, ivermektin gibi anti-solucan ilaçlarıyla tedavi
edilir . Strongiloides tropikal ve alt tropikal bölgelerde bulunur, aynı
zamanda güney ABD dahil ılıman bölgelerde bulunabilir.
Klinik Tanım
Akut enfeksiyon, serpiginous ürtikeryal döküntü, öksürük, nefes darlığı, gastrointestinal semptomlar (ağrı ve yumuşak dışkı dahil) ve kutanöz, alerjik belirtiler ile karakterizedir; ancak enfekte olmuş kişilerin yarısından fazlası asemptomatik kalmaktadır. HTLV1 enfeksiyonu gibi alta yatan başka hastalığı olan veya kortikosteroidler veya immünosüpresif tedavi gören hastalarda Strongyloid hiperinfeksiyon sendromu (SHS) ortaya çıkabilir ve sıklıkla sepsis, şok ve akut solunum sıkıntısı sendromu ile sonuçlanır. Solunum, gastrointestinal, kutanöz ve nörolojik semptomlar değişken yoğunluklarda görülür, ancak SHS’nin ayırt edici özelliği organ yetmezliğinin şiddetidir.
Belirtiler
Akut ve kronik strongyloidiasis asemptomatik
olabilir. Akut strongiloidiazisinin , ilk belirtisi larvanın cilde girdiği
yerinde bir kaşıntı, eritemli döküntü olabilir. Larvalar akciğerlerden ve
trakeadan göç ettikçe öksürük gelişebilir. Gastrointestinal sistemdeki larvalar
ve yetişkin solucanlar karın ağrısı, ishal ve anoreksiye neden olabilir.
Kronik strongyloidiasis , otoenfeksiyon
nedeniyle yıllarca sürebilir. Asemptomatik olabilir veya gastrointestinal,
pulmoner ve / veya kutanöz semptomlarla karakterize edilebilir.
Gastrointestinal şikayetler karın ağrısı ve aralıklı ishal ve kabızlığı içerir.
Açık gastrointestinal kanama oluşabilir ve nadiren dışkıda kan testleri pozitif
olabilir. Semptomlar ülseratif kolit , diğer kronik malabsorpsiyon nedenleri
veya duodenal tıkanıklığın semptomları ile devam edebilir .Ağır enfeksiyonlarda,
otoenfekte larvalar akciğerlerden geçerken öksürük, hırıltılı solunum ve
eozinofili ile Löffler sendromuna neden olabilmesine rağmen, akciğer
semptomları nadirdir . Semptomlar alerjik astım veya kronik obstrüktif akciğer
hastalığına (KOAH) yönelebilir.
Patofizyolojisi
Strongyloides erişkin kurtları, duodenum ve
jejunumun mukozasında ve submukozasında yaşar. Serbest bırakılan yumurtalar,
rabditiform larvaları serbest bırakarak bağırsak lümeninde yumurtadan çıkarlar.
Larvaların çoğu dışkıda atılır. Toprakta birkaç gün sonra, bulaşıcı filariform
larvalara dönüşürler. Kıl kurtları gibi , Strongyloides larvaları insan derisinden
girebilir, kan dolaşımı yoluyla, kılcal damarlardan geçerek, solunum yollarından
akciğerlere ulaşabilir, yutulabilir ve yaklaşık 2 hafta içinde olgunlaşarak
bağırsağa ulaşır. Toprakta, insanlarla temas etmeyen larvalar, larvaları insan
bir konakçıya tekrar girmeden önce birkaç nesil üreyebilen serbest yaşayan yetişkin
solucanlara olgunlaşır.
Epidemiyolojisi
Dünya çapında 30 ila 60 milyon insanı
etkilemektedir ve Afrika, Batı Hint Adaları, Orta ve Güney Amerika, Hint
Okyanusu bölgesi, Güney Doğu Asya gibi alt tropikal bölgelere endemiktir.
Etolojisi
2.5 mm uzunluğunda olan dişi nematodlar,
insanların ince bağırsağında yaşar. Yumurtalar, larvaların dışkılarla atılması
için ince bağırsağın kapağına yerleştirilir. Nemli zeminde, bu larvalar
bulaşıcı aşamalarına doğrudan veya cinsel üreme aşamasından sonra ulaşır. Bu
bulaşıcı formda cilde doğrudan nüfuz edebilirler. Bulaşıcı aşamaya doğru evrim,
bazı durumlarda gözlenen uzun parazitoz süresini (30 yıldan fazla) açıklayan
sindirim sistemi içinde de gerçekleşebilir.
Teşhis
Dışkı veya duodenal dahil örneklerin mikroskobik incelemesi ve
hiperinfeksiyon sendromunda bronşiyal yıkama, balgam veya diğer vücut
sıvılarıyla larvaların tanımlanması
Antikorlar için enzim-immüno analizi
Tek bir dışkı örneğinin mikroskobik
incelemesi, komplike olmayan Strongyloides enfeksiyonlarının yaklaşık% 25’inde
larvaları tespit eder . Konsantre dışkı örneklerinin tekrar tekrar incelenmesi
hassasiyeti artırır; en az 3 ve en fazla 7 dışkı örneği önerilir. Özel dışkı
inceleme yöntemleri hassasiyeti arttırır. Bunlar arasında besin agar plaka
kültürü, Baermann huni tekniği ve Harada-Mori filtre kağıdı tekniği
bulunmaktadır.
Hiperinfeksiyon sendromunda filariform larvalar
dışkı, duodenal içerisinde, balgam ve bronşiyal yıkamalarda ve nadiren beyin
omurilik sıvısında (BOS), idrar, plevral veya asit sıvısında bulunabilir.
Akciğer dokusunun biyopsilerinde veya diğer organların dokularında da görülebilirler.
Akciğer x-rayleri yaygın interstisyel infiltratlar, konsolidasyon veya apse
gösterebilir.
Serumdaki anti-strongiloid antikorlarını
tanımlamak için birkaç bağışıklık tanısı testi mevcuttur. Daha büyük
hassasiyeti ( > % 90) nedeniyle enzim immünolojik testi (EIA) önerilir .
Serum IgG antikorları yaygın yayılmış strongiloidiazisi olan immün sistemi
baskılanmış hastalarda bile tespit edilebilir, ancak saptanabilir antikorların
yokluğu enfeksiyonu dışlamaz. Filariasis veya diğer nematod enfeksiyonları olan
hastalarda çapraz reaksiyonlar yanlış pozitif testlere neden olabilir. Antikor
testi sonuçları, anlık enfeksiyonu geçirilmiş enfeksiyondan ayırt etmek için
kullanılamaz. Pozitif bir test, parazitolojik tanı koymaya yönelik çabaların
sürdürülmesini gerektirir.
Serolojik izleme faydalı olabilir çünkü
başarılı kemoterapiden sonraki 6 ay içinde antikor seviyeleri düşer.
S. stercoralis tanısı için polimeraz zincir
reaksiyonu (PCR) tabanlı yöntemler geliştirilmektedir.
Eozinofili sıklıkla bulunur, ancak
kortikosteroidler veya sitotoksik kemoterapötik ilaçlar gibi ilaçlar tarafından
bastırılabilir.
Tedavi
Ivermektin
Alternatif olarak, albendazol
Strongiloidiazisi olan tüm hastalar tedavi
edilmelidir. İvermektin ile tedavi oranı albendazole göre daha yüksektir.
Ivermectin 200 mcg / kg, 2 gün boyunca günde
bir kez oral olarak, karmaşık olmayan enfeksiyon için kullanılır ve genellikle
iyi tolere edilir. İvermektin ile tedaviden önce hastalar, Loa Loa endemiğinin
bulunduğu Afrika’nın merkez bölgelerine seyahat etmişlerse, Loa Loa tanı testi
uygulamalıdır çünkü ivermektin , loiasis ve yüksek mikrofilaryal hastalarda
ciddi reaksiyonlara neden olabilir. Albendazol 400 mg, 7 gün boyunca günde iki
kez oral yoldan, kuvvetliloidiazisin tedavisi için bir alternatiftir.
İmmün sistemi baskılanmış hastalar balgam ve /
veya dışkı 2 hafta boyunca negatif olana kadar uzun süreli tedavi gerektirir.
Bazen tekrarlanan tedavilere ihtiyaç vardır. Oral ilaç verilemeyen ağır
hastalarda, ivermektinin rektal preparatları veya veteriner deri altı formülasyon
ivermektini kullanılmıştır.
Strongiloidiazili hastalarda hiperinfeksiyon
sendromu yaşamı tehdit eden bir tıbbi acil durumdur. İbitmektin 200 mcg / kg
oral olarak günde bir kez rahabditiform ve filariform larvalar için balgam ve
dışkı incelemeleri 2 hafta boyunca negatif olana kadar devam eder. Geniş
spektrumlu antibiyotikler, bağırsaktan larva istilası ile ilişkili eş zamanlı
polimikrobiyal bakteriyel enfeksiyonları tedavi etmek için kullanılır.
Strongyloidiasis tedavisinden sonra, 2 ila 4
hafta sonra tekrarlanan dışkı muayeneleri ile tedavi belgelenmelidir. Dışkı
pozitif kalırsa, yeniden tedavi belirtilir.
Prognoz
Tedavi edilmezse yaşam boyu enfeksiyon
mümkündür. Çoğu hasta kronik hastalıklarda bile asemptomatik kalır, ancak
prognoz komplikasyonların gelişimine bağlıdır. Yaygın enfeksiyon vakaların%
60-70’inde ölümcüldür.
Anjiostrogylosis,
(sıçan akciğer kurdu enfeksiyonu) endemik olarak görülen zoonotik gıda kaynaklı,
hatta yıkanmamış meyve ve sebzeleri tükettikten sonra ortaya çıkabilen bir
hastalıktır.
Güneydoğu
Asya ve Pasifik Adaları Angiostrongylus pulmoner nematodunun neden olduğu cantonensis.
Enfeksiyon, sebzelerde bulunan bulaşıcı larvaların yutulması veya çiğ ve az
pişmiş salyangozlarda, midye salyangozlarında, kara yengeçlerinde, tatlı su
yengeçlerinde karides, kurbağa ve kertenkele yenilmesiyle ilişkilidir.
Enfeksiyonun ana özelliği eozinofilik menenjittir; ateş, baş ağrısı, halsizlik,
yorgunluk, rinit ile klinik olarak kendini gösteren serebral, bulanık ve çift
görme, öksürük, boyun tutulması, enterit ve kabızlık nematodların
bağırsaklardan akciğerlere, merkezi sisteme hareketinden kaynaklanan
parasteziler sinir sistemi ve gözlere ulaşarak belirtileri başlatır. Şiddetli,
tedavi edilmeyen vakalarda koma ve ölüm meydana gelebilir. Sıklıkla enfeksiyon,
tedavi veya ciddi sonuçlar olmaksızın düzelir, ancak ağır parazit yükü olan
durumlarda, enfeksiyon çok şiddetli olabilir.
Genetik Değişiklikler/Etken Faktörler
A.cantonensis’in birçok
vektörü vardır, en yaygın olanı , Pasifik adalarındaki dev Afrika
kara salyangozu ( Achatina fulica ) ve Tayland ve
Malezya’daki Pila cinsinin salyangozları dahil olmak üzere
birkaç salyangoz türüdür . Altın elma salyangozu A.
canaliculatus , Çin’deki en önemli vektördür. Tatlı su karidesleri,
yengeçler veya diğer paratenik veya taşıma konakları da vektör olarak hareket
edebilir. İnsanlar tesadüfi konakçılardır; larvalar insanlarda çoğalamaz
ve bu nedenle insanlar A. cantonensis yaşam
döngüsüne katkıda bulunmazlar.
Sıçan akciğer kurdu olan nematod
(yuvarlak kurt) Angiostrongylus
cantonensis , insan eozinofilik menenjitinin en yaygın
nedenidir.
Belirti ve Semptomlar
Enfeksiyon ilk önce yavaş
yavaş yükselerek ilerleyen ateş, şiddetli karın ağrısı, bulantı, kusma ve halsizlik,
daha sonra merkezi sinir sistemi (CNS) semptomları ve boynun şiddetli baş
ağrısı ve sertliği ile kendini gösterir. CNS semptomları hafif bilişsel
bozukluk ve yavaş reaksiyonlarla başlar ve çok şiddetli bir formda genellikle
bilinç kaybına ilerler.
Göz istilası belirtileri
arasında görme bozukluğu, ağrı, keratit ve retina
ödemi bulunur . Solucanlar
genellikle ön odada ve vitrözde görülür ve
bazen cerrahi olarak çıkarılabilir.
Kuluçka süresi :
İnsanlarda kuluçka dönemi
enfeksiyondan sonra genellikle 1 haftadan 1 aya kadardır ve 47 güne kadar
uzayabilir. Bu aralık değişir, çünkü insanlar ara konakçıdır ve yaşam
döngüsü bir sıçanda olduğu gibi tahmin edilebilir şekilde devam etmez.
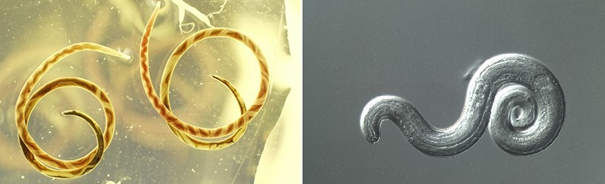
Görsel 1, kaynak: Sol: Sıçan akciğerlerinden iki Angiostrongylus erişkin kadın iyileşti. Her iki solucanda görülen ayırt edici, sarmal desen beyaz uterus tüpleri ve kırmızı, kan dolu bağırsak tarafından yaratılır. Sağ: Angiostrongylus cantonensis üçüncü evre (L3), bir sümüklübeden enfektif larva. Diferansiyel girişim kontrast (DIC) mikroskobu altında yakalanan görüntü.
Genetik Görülme Sıklığı
Genetik
bir bilgi bulunmamaktadır. A. cantonensis ve vektörleri Güneydoğu Asya ve
Pasifik Havzası’na endemiktir. Küreselleşme gittikçe daha fazla yere
yayılmasına ve daha fazla gezgin parazitlerle karşılaştıkça enfeksiyon giderek
daha önemli hale geliyor. Parazitler muhtemelen gemilerde kaçak yol olarak
seyahat eden sıçanlardan ve endemik alanların dışında salyangoz vektörlerinin
sokulmasıyla etkili bir şekilde seyahat ederler.
Parazit
endemik bölgelerin dışında nadiren görülür ve bu durumlarda hastalar genellikle
endemik bir bölgeye seyahat öyküsü vardır.
Angiostrongyliasis’in
teşhisi, angiostrongylus larvalarının kendilerini
gösterme zorluğu nedeniyle karmaşıktır ve genellikle eozinofilik
menenjitin varlığına ve salyangoz konakçılarına maruz kalma öyküsüne dayanarak
yapılır. Eozinofilik menenjit genellikle BOS’ta > 10 eozinofil / μL veya toplam BOS lökosit sayısında en
az% 10 eozinofil içeren bir menenjit olarak karakterize
edilir . Beyin omurilik sıvısında bulunan veya cerrahi olarak gözden
çıkarılan solucanlar Angiostrongyliasis’i teşhis etmek için tanımlanabilir.
Lomber ponksiyon her zaman
menenjit şüphesi olan durumlarda yapılmalıdır. Eozinofilik menenjit
vakalarında, BOS’ta bulunduklarında bile nadiren solucanlar üretecektir, çünkü
sinirlerin sonuna yapışmaya eğilimlidirler.
BT veya MRG’de hem gri hem
de beyaz cevher invazyonlu beyin lezyonları görülebilir. Bununla birlikte,
MRG bulguları sonuçsuz olma eğilimindedir ve genellikle spesifik olmayan
lezyonlar ve ventriküler genişlemeyi içerir. Bazen göç eden solucanlar
tarafından üretilen bir kanama vardır ve teşhis değeri vardır.
Yüksek eozinofilleri olan
hastalarda seroloji, başka bir parazit ile enfeksiyondan ziyade
Angiostrongylias tanısını doğrulamak için kullanılabilir.
En kesin tanı her zaman BOS
veya gözde bulunan larvaların tanımlanmasından kaynaklanır, ancak bu nadirlik
nedeniyle yukarıdaki testlere dayanan bir klinik tanı büyük olasılıkla vardır.
Tedavi
Angiostrongiliyazis için
gelişme aşamasında hiçbir aşı yoktur.
Bireyler için öneriler
Endemik bölgelerde enfeksiyondan
kaçınmak için ;
Salyangoz ve tatlı su karidesleri gibi pişmemiş vektörlerin tüketiminden
kaçının
Vektörlerle kirlenmiş olabilecek açık kaynaklardan içme suyundan kaçının
Küçük çocukların canlı salyangozlarla oynamasını veya yemesini önleyin
Anjiyostrongiliyazis
tedavisi iyi tanımlanmamıştır, ancak çoğu strateji solucanları öldürmek için
anti-parazitiklerin, solucanlar ölürken iltihabı sınırlayan steroidlerin ve
menenjit semptomlarını yönetmek için ağrı kesici ilaçların bir kombinasyonunu
içerir.
Semptomatik tedavi, bulantı,
kusma, baş ağrısı ve bazı durumlarda sinir hasarı veya kas atrofisine bağlı
kronik ağrı gibi belirtiler için endikedir.
İnsanlara yapılan en iyi
öneri, yiyeceğiniz çiğ ürünleri iyice yıkayın, ellerinizi yıkayın, salyangoz
veya sümüklü böcek veya fare yemeyin.
Gorham-Stout hastalığı
(kaybolan kemik hastalığı) (GSD), lenfatik damarların çoğalması ve genişlemesi
ile ilişkili nadir bir masif osteoliz hastalığıdır. GSD vücuttaki herhangi bir
kemiği etkileyebilir ve monostotik veya poliostotik olabilir. Tıp literatüründe
kaybolan kemik hastalığı, masif osteoliz olarak da bilinen Gorham-Stout
hastalığı (GSD), ilerleyici kemik kaybı (osteoliz) ile karakterize nadir bir
kemik bozukluğudur ve lenfatik damarların aşırı çoğalması (proliferasyonu).
Etkilenen bireyler, ilerleyen kemik yıkımı ve emilimine maruz kalırlar. Birden
fazla kemik tutulabilir.
GSD her yaşta ortaya
çıkabilir, ancak genellikle çocuklarda ve genç yetişkinlerde (ortalama 13 yıl)
teşhis edilir. GSD vücuttaki herhangi bir kemiği etkileyebilir, ancak en yaygın
olarak kaburgaları, ardından kafatası, klavikula ve servikal omurga etkiler.
Etkilenen diğer alanlar arasında maksillofasiyal kemikler (çoğunlukla çene
kemiği), sternum, humerus, el, femur ve ayak bulunur. GSD monostotik veya
poliostotik olabilir ve semptomlar etkilenen vücut bölgelerine göre değişir. En
yaygın semptom lokalize ağrıdır. Etkilenen uzuvların şişmesi, zayıflığı ve
fonksiyonel bozukluğu da fark edilir. Dentoalveoler bölgede hareketli dişler,
malokluzyon, mandibular sapma ve kemik deformitesi görülebilir. Torasik
tutulumu olan hastalar solunum sıkıntısı (şilotoraksın neden olduğu)
gösterebilir. Vertebral tutuluma sekonder şiddetli nörolojik defektler ve felç
de gözlenir. Servikal omurga veya kafatasının hastalığı olan hastalarda beyin
omurilik sıvısı sızıntısı gelişebilir. GSD, bir kemik kırılmasından sonra
(spontan veya küçük travmadan sonra) bulunabilir.
GSD etiyolojisi hala anlaşılması güçtür.
Patolojik süreç, kemiğe bitişik veya kemik içindeki endotel kanallarının iyi
huylu vasküler proliferasyonu olup kemik trabekülünün aşırı incelmesine,
osteoklast aracılı rezorpsiyona ve kemiğin fibröz doku ile değiştirilmesine yol
açar. Doku örnekleri lenfatik endotel hücre markerleri için pozitif test eder,
bu da GSD’nin düzensiz lenfanjiyogenez içeren bir hastalık olduğunu düşündürür.
Kemik kaybı (osteoliz) enfeksiyon,
inflamasyon, kanser ve bazı endokrin bozuklukları içeren çeşitli farklı
durumlardan kaynaklanabilir. GSD’nin ayırıcı tanısında Hajdu-Cheney sendromu,
Paget hastalığı, romatoid artrit, fibröz displazi, Langerhans hücre
histiyositozu, Winchester sendromu, karpal tarsal osteoliz, idiyopatik
multisentrik osteoliz, nefropatili multisentrik osteoliz ve eozopozizofizozit
yer alır . (Bu bozukluklar hakkında daha fazla bilgi için, Nadir Hastalık
Veritabanında arama teriminiz olarak belirli bir bozukluk adı seçin.)
Genelleştirilmiş lenfatik anomali (daha
önce lenfanjiyotoz olarak bilinen GLA) GSD ile yakından ilişkilidir. GLA
hastaları multifokal lenfatik malformasyonlara sahiptir. Bu malformasyonlar
kemikte mevcut olabilir, ancak GSD’de görüldüğü gibi kortikal kemik kaybına
neden olmaz.
Belirti ve Semptomlar
Gorham’ın yok olan kemik
hastalığı ve fantom kemik hastalığı olarak da bilinen Gorham hastalığı
(belirgin GOR-amz), içerisindeki bozuk, ince duvarlı vasküler veya lenfatik
kanalların kontrolsüz çoğalmasıyla karakterize, çok nadir bir iskelet
durumudur. kemiğin emilimine ve kemiğin anjiyomlar ve / veya fibroz ile değiştirilmesine
yol açan kemik hastalığı. GSD’den yaygın olarak etkilenen bölgeler arasında
kaburgalar, omurga, pelvis, kafatası, köprücük kemiği (klavikula) ve çene
bulunur. Etkilenen bölgede ağrı ve şişme oluşabilir.
Bazı durumlarda,
kendiliğinden veya düşme gibi küçük travmayı takiben bir kırık oluşana kadar
hiçbir belirti görülmez. Akut bir lokalize ağrı ve şişme oluşabilir. Daha
yaygın olarak, belirgin bir nedeni olmayan ağrı, zaman içinde frekans ve
yoğunlukta artar ve sonunda alanın zayıflığı ve belirgin deformitesi ile
birlikte olabilir. İlerleme oranı tahmin edilemez ve prognoz zor olabilir.
Hastalık birkaç yıl sonra stabilize olabilir, spontan remisyona girebilir veya
göğüs ve üst omurga içeren vakalarda ölümcül olabilir. Remisyondan sonra
hastalığın tekrarlaması da ortaya çıkabilir. Omurga ve kafatası tabanının
tutulması nörolojik komplikasyonlardan kötü bir sonuca neden olabilir. Birçok
durumda, Gorham hastalığının sonucu ciddi deformite ve fonksiyonel sakatlıktır.
Hastalık kaburgalarda,
skapulada veya torasik omurlarda mevcutsa nefes almada zorluk ve göğüs ağrısı
gibi belirtiler görülebilir. Bunlar, hastalığın kemikten göğüs boşluğuna
yayıldığını gösterebilir. Solunum problemleri astım olarak yanlış teşhis
edilebilir, çünkü akciğerlere verilen hasar, astımda görülen akciğer fonksiyon
testinde aynı tür değişikliklere neden olabilir. Lezyonların göğse uzatılması,
şilöz plevral ve perikardiyal efüzyonların gelişmesine yol açabilir. Chyle,
enfeksiyonla mücadelede önemli olan protein ve beyaz kan hücreleri bakımından
zengindir. Chyle’nin göğse kaybolması, enfeksiyon, yetersiz beslenme ve solunum
sıkıntısı ve başarısızlığı gibi ciddi sonuçlara neden olabilir. Bu
komplikasyonlar veya solunum güçlüğü, göğüs ağrısı, zayıf büyüme veya kilo
kaybı ve enfeksiyon gibi semptomları bazen durumun ilk belirtileri olmuştur.
Gorham hastalığının spesifik
nedeni bilinmemektedir. 1990’lı yıllardan başlayarak, hastalığı olan kişilerde
interlökin-6 (IL-6) adı verilen bir proteinin yüksek seviyelerinin tespit
edildiği bildirildi ve bu da bazılarının artmış IL-6 ve vasküler endotelyal
büyüme faktörünün (VEGF) seviyelerinin katkıda bulunabileceğini düşündürdü.
1999 yılında Möller ve
arkadaşları, “Gorham-Stout sendromu, esasen, artan sayıda parakrin veya otokrin
ile uyarılan hiperaktif osteoklast nedeniyle ciddi bir şekilde artmış kemik
rezorpsiyonu olan monosentrik bir kemik hastalığı olabilir. osteoklastların
varlığı veya yokluğu veya osteoklast sayısı ile ilgili belirgin çelişki,
sendromun farklı evreleri ile açıklanabilir. ” Ayrıca histopatolojik
çalışmalarının Gorham hastalığında görülen osteolitik değişikliklerin
hiperaktif osteoklastik kemiğin sonucu olduğuna dair iyi kanıt sunduğunu
belirtmişlerdir. Bununla birlikte, diğerleri lenfanjiyotoz ve Gorham
hastalığının ayrı hastalıklardan ziyade bir hastalık spektrumu olarak
değerlendirilmesi gerektiği sonucuna varmıştır.
Gorham’ın dengesiz
osteoklastik aktiviteden kaynaklandığına dair bir fikir birliği olsa da, bu
davranışın başlamasına neden olan şey hakkında kesin bir kanıt bulunamamıştır.
Kalıtım Paterni/Deseni
Bugüne kadar literatürde
yaklaşık 300 vaka bildirilmiştir. GSD açık bir ırk, cinsiyet tercihi (1.6: 1;
erkek: kadın oranı) veya coğrafi dağılım göstermemektedir. GSD’nin kesin nedeni
bilinmemektedir. GSD çok nadir
olduğu için, birçok vaka teşhis edilmez veya yanlış teşhis edilir, bu da
bozukluğun genel popülasyondaki gerçek sıklığını belirlemeyi zorlaştırır.
Teşhis ve Tedavi Yöntemleri
Teşhis
Tanı progresif osteolizi ve
kortikal yıkımı gösteren radyografik bulgulara dayanır. Manyetik rezonans
görüntüleme, kemikte tam rezorpsiyon ve T1 ağırlıklı görüntülemede düşük sinyal
yoğunluğu ve T2’de yüksek sinyal yoğunluğu olan ve kontrast görüntülemede yoğun
artış gösteren infiltratif yumuşak doku ile yer değiştirmeyi gösterir. Lenfatik
endotelyal hücrelerin (LYVE-1, podoplanin / D2-40) immünohistokimyasal
belirteçleri, kemiklerin medüller ve kortikal bölgelerinde ve etkilenen yumuşak
dokularda lenfatik damarların varlığını ortaya çıkarır. Kaburga lezyonları
biyopsi yapılmamalıdır, çünkü bu prosedür refrakter bir şilöz efüzyon ortaya
çıkarabilir.
Tedavi
GSD’nin tedavisi, ilerleyici
hastalığı stabilize etmek için ilaçları (bisfosfonatlar ve / veya interferon
alfa 2b, sirolimus da araştırılmaktadır) ve şilotoraksı azaltabilen veya
durdurabilen destekleyici prosedürleri (plörektomi, plörodez, torasentez ve
torasik kanal embolizasyonu veya ligasyonu) içerebilir veya iskeletin etkilenen
bölgelerini stabilize edebilir. Radyoterapi bu terapilerle kombinasyon halinde
kullanılabilir, ancak genellikle refrakter veya hızla ilerleyen hastalıklara
ayrılır.
Prognoz, etkilenen alanların
boyutuna ve yerine bağlıdır. Hafif hastalık yıllarca stabil kalabilirken,
kraniyofasiyal ve / veya torasik bölgeleri içeren ciddi vakalar ölümcül
olabilir. Akciğer tutulumu kötüleşen bir prognozun göstergesidir.
Gorham hastalığının tedavisi
çoğunlukla palyatiftir ve semptom yönetimi ile sınırlıdır.
Bazen kemik yıkımı
kendiliğinden sona erer ve tedavi gerekmez, ancak hastalık ilerlediğinde
agresif müdahale gerekebilir. Duffy ve meslektaşları, kaburga, omuz veya üst
omurgada Gorham hastalığı olan kişilerin yaklaşık% 17’sinin, hastalığın göğsüne
genişlemesini yaşadığını ve ciddi sonuçlarıyla şilotoraksa yol açtığını ve bu
gruptaki ölüm oranının cerrahi müdahale olmadan% 64’e kadar çıkabilir.
Şilomikron Retansiyon Hastalığı besinlerden yağ, kolesterol
ve bazı vitaminlerin alımının bozulduğu kalıtsal bir hastalıktır. Başlıca
sindirim sistemini ve sinir sistemini etkiler. Bu hastalık bebeklik ya da erken
çocukluk çağında ortaya çıkar. Etkilenen çocuklarda kandaki kolesterol seviyesi
düşüktür. Ayrıca E vitamini eksikliği, büyüme geriliği, sürekli ishal, kötü
kokulu dışkı görülür.
Belirti ve
Semptomlar
Hastaların tümünde kolesterol düşüklüğü ve ishal görülür.
Bunların yanında bireylere göre çeşitlilik gösteren birçok belirti vardır.
Yüksek seviyede karaciğer enzimleri, retina hastalıkları, dışkıda yağ fazlalığı
(steatore), şişkinlik, vitamin metabolizması bozuklukları da etkilenen
bireylerde görülen belirtilerdendir. Çocukluk çağının sonlarında sinir
sisteminde bozukluklar da gelişebilir.
Görülme
Sıklığı
Dünya çapında bugüne kadar yaklaşık 55 vaka tanımlanmıştır.
Genetik
Değişiklikler
SAR1B genindeki
mutasyonlar Şilomikron Retansiyon Hastalığına neden olmaktadır. SAR1B geni Sar1b proteinini kodlar. Bu
protein şilomikron denen moleküllerin taşınmasında rol oynar. Şilomikronlar
sindirim sırasında besin öğelerinin emilimini sağlayan ve yağların, yağda
çözünen vitaminlerin ve kolesterolün ince bağırsaktan kana taşınmasını sağlayan
moleküllerdir. SAR1B geninde meydana
gelen mutasyonlar sonucu bu besin maddelerinin emilimi ciddi ölçüde
azalmaktadır.
Kalıtım
Paterni
Bu hastalık otozomal resesif (çekinik) kalıtılır. Yani
hastalığın oluşması için her iki gen kopyasında da mutasyon olması gerekir.
Teşhis
Yöntemleri
Genelde spesifik semptomlar olmadığı için hastalığın teşhisi
zordur. Teşhis, hastada görülen kronik ishal, yağ emilim bozukluğu ve anormal lipit
profiline (normal trigliserit varlığında, yaklaşık %50 düşük kolesterol) göre
konulabilir. Genetik tanı testleri ile SAR1B
genindeki mutasyonun belirlenmesi mümkündür.
Tedavi
Yöntemleri
Tedavi yaklaşımı beslenme ve büyümenin takibine yönelik
olmalıdır. Uygulanacak tedavi, semptomların belirlenmesi ve önlenmesine
odaklanır. E vitamini eksikliğinin kontrolü nörolojik problemlerin ortaya
çıkmaması için ciddi derecede öneme sahiptir. Tedavi, yağda çözünen vitaminler
ve büyük miktarda E vitamini takviyesi içerir. Diğer belirtilerin önlenmesi
için de yağ alımının çok sıkı takibini içeren özel diyetler uygulanmaktadır.
Hastalığın
Diğer İsimleri
CMRD, Anderson Sendromu, Bağırsakta yağ emilim bozukluğu,
Bağırsak hücrelerinde Hipobetalipoproteinemi’yle birlikte apolipoprotein b
benzeri protein birikimi
Genel
Bilgi, Genetik Değişiklikler/ Etken Faktörler
Proksimal
spinal müsküler atrofiler, omurilik ve beyin sapı çekirdeğindeki alt motor
nöronların dejenerasyonu ve kaybından kaynaklanan ilerleyici kas güçsüzlüğü ile
karakterize bir grup nöromüsküler bozukluktur. Hastalığın başlangıç yaşı ve
şiddetine göre dört alt tip tanımlanmıştır: altı aydan önce başlayan en
şiddetli form olan tip 1 (SMA1); 6 ila 18 aylık arasında başlayan tip 2 (SMA2),
çocukluk ve ergenlik arasında başlayan tip 3 (SMA3) ve yetişkin başlangıcında
en az şiddetli olan tip 4 (SMA4). Tüm tipler, özellikle alt ekstremite ve
solunum kaslarını etkileyen kas zayıflığı ve değişen şiddette atrofi ile
karakterizedir. Zayıflık neredeyse her zaman simetrik ve ilerleyicidir.
Skolyoz, kas retraksiyonları ve eklem kontraktürleri ortaya çıkabilir. Kabızlık
ve gastroözofageal reflü sık görülür.
Hastalık
belirgin olarak SMN proteini sentezleyen SMN1 ve SMN2 geninde gerçekleşen
mutasyonlardan kaynaklanır ve SMN proteini üretemezler. Bunun sonucunda motor
nöron sinirleri beslenemez hale gelir ve istemli kasların çalışmasında sorunlar
meydana gelmektedir.
Belirti
ve Semptomlar
Kişiden kişiye
değişebileceği gibi genelde görülen belirtiler aşağıdaki gibidir.
Kas güçsüzlüğü,
Zihinsel gerilik
Tendon reflekslerinde gerileme
Mikrosefali (baş ve çevresinin
normalden küçük olması durumu)
Alt ekstremite amyotrofisi
(çizgili kas telinin yok olmasıyla sonuçlanabilen hacim azalması)
Skolyoz
Hemiparezi/ kısmi felç
Solunum güçlüğü
EMG anormalliği
–elektromiyografi (EMG) kas hastalığının, hasar bulunan sinirin tespitinde kullanılan
görüntüleme yöntemi
Kabızlık, gasroözofageal reflü
Genetik
Görülme Sıklığı
Her 30.000 kişiden 1’inde görülüyordur.
Kalıtım
Paterni/ Deseni
Otozomal
resesif olarak kalıtılır. Bu sebeple hasta bireyin biyolojik anne ve babasında
mutasyona uğramış ilgili genin bir kopyası taşınıyor demektir. Taşıyıcı olan
ebeveynler hasta değillerdir veya herhangi bir belirti göstermezler.
Teşhis
Yöntemleri ve Tedaviler
Teşhis ve tanı
hastanın tıbbi özgeçmişi ve muayeneye dayanmaktadır. Aynı şekilde yapılan
genetik testlerle doğrulanabilmektedir. Elektromiyografi (EMG) kas
hastalığının, hasar bulunan sinirin tespitinde kullanılan görüntüleme yöntemi
ve kas biyopsisi yapılabilmektedir. Ayırıcı teşhis arasında amiyotrofik lateral
skleroz, konjenital kas distrofileri, konjenital miyopatiler, primer lateral
skleroz, miyastenia gravis ve karbonhidrat metabolizması bozuklukları bulunabilmektedir.
Doğum öncesi tanı ise amniyositlerin veya koryonik villus örneklerinin
moleküler analizi ile mümkündür.
Tedavi olarak
çoğunluğu semptomatik olmak üzere birkaç yöntem bulunmaktadır.
Spiraza,
SMA
hastaları, SMN1 genindeki bir mutasyon nedeniyle hayatta kalma motor nöronu
(SMN) adı verilen bir proteini üretmezler. Spinraza daha az etkili ikinci bir
gen olan SMN2’nin daha fonksiyonel SMN proteini üretme yeteneğini arttırır. Vücuttaki
SMN proteini miktarını arttırarak, Spinraza motor nöron ölümünü geciktirmeye ve
hastalık semptomlarının ilerlemesini yavaşlatmaya yardımcı olabilir.
Zolgensma,
SMN1 geninin sağlıklı bir kopyasını hedef motor nöronlarına vermek için genetik
olarak tasarlanmış bir virüs kullanır. 2 yaş ve altındaki SMA1 hastalarına
uygulanan tek seferlik enjekte edilen bir gen terapisi yöntemidir.
SMA’da özel
olarak araştırılmayan diğer tedaviler hastalık semptomlarını yönetmek veya
komplikasyonları önlemek için de kullanılabilir. Örneğin, kasların sert ve
gergin olduğu spastisite, baklofen, tizanidin veya benzodiazepinler gibi kas
gevşeticiler tarafından hafifletilebilir.
Ayrıca hastada
geilşen semptom ve komplikasyonlar fizik tedavi, nefes tedavisi ve beslenme
destekleri ile bunları engelleme ve geciktirmeye yardımcı olmaktadır.
Hastalıkla
İlişkili Genler
SETX, SMNDC1, FBLN5, KIF1B, SPG7, SMN1,
SMN2, APOE, BICD2, DES, DNAI1, AG genleri genel anlamda hastalıktan sorumlu
genler arasında gösterilmektedir. Her tip SMA’da ilişkili genler veya ilişki
oranları değişmektedir. İlgilendiğiniz SMA tipinin sayfasına bakarak ilişkili
geni bulabilirsiniz.
Kol-Kuşak Kas Distrofisi Tip 2D (LGMD2D), kalça ve omuz bölgelerinin (kol-kuşak bölgeleri)
gönüllü kaslarının israfı (atrofi) ve zayıflığı ile karakterize bir nadir hastalıktır. Kas
zayıflığı ve atrofi ilerleyicidir ve vücudun diğer kaslarını etkileyecek kadar
yayılabilir. Semptomlarının başlangıç yaşı, şiddeti ve ilerlemesi, aynı
ailedeki bireyler arasında bile durumdan duruma değişebilir. Bazı bireylerde
hafif, yavaş ilerleyen bozukluklara sahip olabilirken bazı bireylerde şiddetli
sakatlığa neden olabilecek hızla ilerleyen bir bozukluğa sahip olabilir. Bu
bozukluklar artık genetik ve protein analizi ile ayırt edilebilir.
LGMD2D, alfa-sarkoglikan a proteinin geni olan ve
17q21.33’te lokalize olan (konumlanan) SGCA geninde meydana gelen değişimin
(mutasyonun) neden olduğu otozomal resesif bir genetik durumdur. Genelde bireyde çocukluk veya ergenlik çağında başlar ve ilerleyicidir. Çoğu durumda, LGMD’nin çocukluk çağında
başlaması, ergen veya erişkin başlangıçlı vakalardan daha hızlı ilerleyen daha
ciddi bir bozukluğa neden olur.
Kol-Kuşak Kas Distrofisi
Tip 2D, esas olarak proksimal kasları etkilemesi sonucu yürüme zorluğu,
skapular kanatlanma, baldır psödohipertrofisi ve tiptoe yürüyüş gibi sonuçlara neden olur. Kardiyak
ve solunumsal tutulum nadirdir. Kardiyomiyopati de nadiren
bildirilmiştir.
Genetik
Değişiklikler/Etken Faktörler
Kol-Kuşak Kas Distrofisinde etkili olan gen SGCA
(GKRY) genidir. SGCA geni
sarkoglikan protein kompleksi adı verilen bir protein grubunun alfa bileşeni
(alt-birim) yapmak için talimatlar içerir. Sarkoglikan protein kompleksi,
kas hücrelerini çevreleyen zarda bulunur. Distrofinler ve distrolikanlar
adı verilen proteinlerden oluşan distrofin kompleksine yapışarak (bağlanarak)
stabilize ederek (sağlamlaştırarak) kas dokusunun yapısının korunmasına
yardımcı olur. Büyük distrofin kompleksi, kas liflerini güçlendirir ve
kasları yaralanmaya karşı korur. Her kas hücresinin yapısal çerçevesini
(hücre iskeletini) protein kafes ve hücre dışındaki diğer moleküllere (hücre
dışı matris) bağlayan bir çapa görevi görür.
SGCA gen
mutasyonu, protein yapı bloğunun (amino asit) argininini, Arg77Cys veya R77C
olarak yazılan alfa-sarkoglikan proteininde 77. pozisyondaki amino asit sistein
ile değiştirir. Bilinen SGCA gen
mutasyonlarının geri kalanı, bireysel ailelere veya belirli
popülasyonlara özgüdür.
SGCA gen
mutasyonları sarkoglikan kompleksinin distrofin kompleksini oluşturmasını veya
bunstrofin kompleksine bağlanmasını ve stabilize olmasını önleyebilir. Bu
komplekslerle ilgili problemler kas liflerinin gücünü ve esnekliğini azaltarak
Kol-Kuşak Kas Distrofisine neden olur.
Belirti
ve Semptomlar
Bu kısımda Kol-Kuşak
Kas Distrofisi tip 2D hastalığının belirtileri listelenmiştir. Ancak çoğu
hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan
insanlar listelenen tüm semptomlara sahip olmayabilir.
İnsanların %30 -79 bu belirtilere sahiptir:
Aşil tendonu kontraktürü
(kısalması)
Buzağı kas psödohipertrofisi
Merdiven çıkma zorluğu
Yüksek serum kreatin kinaz (dolaşımda yüksek
kreatin fosfokinaz enzim düzeyi)
Sık düşme
Bu belirtilerin yanında skapular kanatlanma, hiperlordoz, sınırlı omuz hareketi, proksimal kas güçsüzlüğü, ayak yürüyüşü, vatka yürüyüşü gibi belirtiler de görülür. Nadir olarak torasik skolyoz da gözlenmiştir.
LGMD2D’nin klinik spektrumu 10-12 yaşlarında
ambulasyon (ayağa kalkma, yürüme) kaybına yol açan erken başlangıçlı ve hızlı
ilerleyen hastalar (a), ergenlikte yavaş
ilerleyen çocukluk başlangıçlı formları (b, c) ve yetişkinlikte başlayan
miyopatik değişikliklere sahip daha hafif formlar (d)
Kol-Kuşak Kas Distrofisinin prevalansını belirlemek zordur, çünkü özellikleri değişir ve
diğer kas bozukluklarınınki ile çakışır. Prevalans tahminleri 14.500’de 1
ile 123.000 kişide 1 arasında değişmektedir.
Kalıtım
Paterni
Kol-Kuşak Kas Distrofisi Tip 2D, otozomal resesif bir
paternde kalıtsaldır. Yani her bir
hücredeki genin her iki kopyasının mutasyonları vardır. Resesif genetik bozukluklarda hasta birey, her bir
ebeveynden aynı özellik için aynı anormal geni miras aldığında ortaya
çıkar. Bir kişi hastalık için bir normal gen ve bir gen alırsa, kişi hastalık
için bir taşıyıcı olacaktır ancak genellikle hastalığın belirtilerini göstermez. İki
taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip
olma riski her hamilelikte % 25’tir. Her hamilelikte ebeveyn gibi taşıyıcı
olan bir çocuk sahibi olma riski % 50’dir. Bir çocuğun her iki ebeveynden
de normal gen alma ve bu özellik için genetik olarak normal olma şansı %25’tir. Risk
erkekler ve kadınlar için eşittir.
Teşhis
Yöntemleri
LGMD (Kol-Kuşak Kas
Distrofisi) grubunda, hastaya ve ailesine doğru genetik tavsiye verilebilmesi
ve hastalıkların varlığından hastalık varlığına değişebilen komplikasyonların
yönetimi için uygun rehberlik sağlanabilmesi için kesin bir tanıya ulaşmak
önemlidir. Bu özellikle kardiyak veya solunumsal komplikasyon riski ile
ilgilidir. Bugün mevcut olan kesin test, geçmişte LGMD tanısı konan
bireylerin yeniden değerlendirilmesini ve daha kesin bir moleküler tanı
verilmesini mümkün kılabilir.
LGMD tanısı, kapsamlı bir
klinik değerlendirme, ayrıntılı bir hasta öyküsü, karakteristik semptomların
tanımlanması (örn. kas zayıflığı ve atrofinin spesifik dağılımı) ve cerrahi
olarak çıkarılması ve mikroskopik muayenesi (biyopsi) dahil olmak üzere çeşitli
özel testlere dayanarak konur. Bu özel testlere kasların sağlığını ve kasları
kontrol eden sinirleri değerlendiren bir test olan elektromiyografi, özel
kan testleri, belirli kas proteinlerinin varlığını ve sayısını değerlendiren
testler (immünohistokimya) ve moleküler genetik test örenk verilebilir.
Bir elektromiyografi
sırasında, bir iğne elektrodu deriden etkilenen bir kas içine
sokulur. Elektrot kasın elektriksel aktivitesini kaydeder. Bu kayıt,
bir kasın sinirlere ne kadar iyi tepki verdiğini gösterir ve kas zayıflığının
kasın kendisinden mi yoksa kasları kontrol eden sinirlerden mi kaynaklandığını
belirleyebilir. Elektromiyografi testi, belirli bir LGMD alt tipinin
teşhisine izin vermez, ancak alternatif teşhisleri dışlamak için yararlı
olabilir.
Kan testleri, kas hasar
gördüğünde anormal derecede yüksek seviyelerde bulunan bir enzim olan yüksek
kreatin kinaz (CK) seviyelerini ortaya çıkarabilir. Yüksek CK seviyeleri,
tüm LGMD vakalarında olmasa da bazılarında görülür. LGMD’nin otozomal
resesif formlarında CK düzeyleri, otozomal dominant formlardan çok daha
yüksektir. Yüksek CK seviyelerinin tespiti, kasın hasar gördüğünü veya
iltihaplandığını doğrulayabilir, ancak LGMD tanısını doğrulayamaz. Bununla
birlikte, hangi tip LGMD’nin diğerlerinden daha olası olduğunu belirtmek
yardımcı olabilir.
Moleküler genetik test,
spesifik bir genetik mutasyonu tanımlamak için deoksiribonükleik asitin (DNA)
incelenmesini içerir. Bu artık LGMD’de tanı için altın standarttır ve
diğer aile üyeleri için spesifik bir tanıya ve spesifik testlere izin verir.
LGMD vakıflarından oluşan bir
konsorsiyum, kas güçsüzlüğü olan hastalara ücretsiz genetik sekans
sunmak için http://LGMD-diagnosis.org adresinde teşhis programı oluşturdu. Kas
güçsüzlükleri için genetik bir açıklaması olmayan bireylerin, serbest genetik
sekanslama için uygun olup olmadıklarını belirlemek için alabilecekleri
çevrimiçi bir test sunmaktadır. Doktorlar, hastalarının uygun olup
olmadığını belirlemek için Jain Vakfı tarafından geliştirilen Otomatik LGMD
Teşhis Asistanını (ALDA) kullanarak hastaları adına da başvurabilirler.
Tedavi Yöntemleri
LGMD’nin herhangi bir formu
için herhangi bir tedavi yoktur. Tedavi, her bir kişide mevcut olan
spesifik semptomlara yöneliktir. Özel tedavi seçenekleri arasında kas
gücünü artırmak ve kontraktürleri (kısalmaları) önlemek için fiziksel ve
mesleki terapi, yürüme (ambulasyon) ve hareketliliğe yardımcı olmak için
çeşitli cihazların (örn. bastonlar, diş telleri, yürüyüşçüler, tekerlekli sandalyeler)
kullanılması ve skolyoz gibi iskelet anormalliklerini düzeltmek için
cerrahi gibi yöntemler yer almaktadır.
Genetik danışmanlık etkilenen
bireyler ve aileleri için yararlı olabilir. Diğer tedaviler semptomatik
(belirtilere yönelik) ve destekleyicidir. Hastalara ilgili hasta
organizasyonları ve kayıtları için iletişim bilgileri verilmelidir.
Hastalıkla
İlişkili Genler
SGCA geni (GKRY)
Hastalığın
Diğer İsimleri
LGMD
Tip 2D
LGMD2D
LGMDR3
Pelvofemoral Kas Distrofisi
Proksimal Kas Distrofisi
Alfa-sarkoglikan ile Kas Distrofisi Uzuv Kemeri
Alfa-sarkoglikan
ile ilgili LGMDR3
Alfa-sarkoglikan
eksikliğine bağlı LGMD
Alfa-sargoglikan
eksikliğine bağlı Ekstremite Kuşak Kas Distrofisi
Alfa-sarcoglycanopathy
Duchenne Benzeri Otozomal Resesif Kas Distrofisi, Tip 2
Alfa-mannosidoz
(α-mannosidoz), lizozomal α-D-mannosidaz enzimini kodlayan gendeki
mutasyonların neden olduğu otozomal resesif kalıtım ile seyrek görülen bir
lizozomal depo hastalığıdır. Zihinsel yetersizlik, işitme kaybı, ataksi,
iskelet anormallikleri ve anormal yüz özellikleri ile karakterizedir. Belirti
ve semptomlar hastalığın şiddetine göre değişiklik gösterebilir. Genellikle
hafif ila orta derecede zihinsel engel, işitme kaybı, zayıflamış bağışıklık
sistemi, ayırt edici yüz özellikleri ve serebellar bozukluklar (örn. , ataksi)
görülür.
Belirti ve Semptomlar
Alfa-mannosidozun
semptomları, ilerlemesi ve şiddeti, aynı mutasyonu paylaşan kardeşler dahil
olmak üzere bir kişiden diğerine büyük ölçüde değişiklik gösterir. Bazı
bireylerde doğumdan kısa bir süre sonra semptomlar gelişebilir. Bebeklik veya
erken çocukluk döneminde potansiyel olarak hayatı tehdit eden komplikasyonlar
gelişebilir. Diğer bireylerde ise genellikle 10 yaşından önce başlayan daha
ılımlı semptomlar gelişebilir. Bazı durumlarda ise bireyler yetişkinliğe kadar
teşhis edilemeyebilir. Genel olarak, etkilenen bireyler zihinsel engel, ayırt
edici yüz özellikleri ve iskelet anormalliklerine sahip olabilir. Semptomlar
zamanla yavaşça kötüleşebilir.
Semptomların
şiddetine göre 3 alt tipte sınıflandırılır:
Tip 1: Kas problemleri (miyopati) ile yavaş ilerlemenin olduğu on yaşından sonra gelişen semptomları içeren hafif bir formdur.
Tip 2: İskelet anormallikleri, miyopati ve yavaş ilerleme ile birlikte on yaşından önce semptomların görüldüğü ılımlı bir formdur. İskelet anormallikleri genellikle azalmış kemik yoğunluğu, kafatasının üstündeki kemiklerin kalınlaşması, omurgadaki kemiklerin deformasyonları, eğilmiş bacaklar, kemik ve eklemlerde deformasyonları içerir. En yaygın olan tiptir.
Tip 3: Progresif santral sinir sistemi tutulumu ve enfeksiyonundan dolayı gebelik kaybı veya erken ölüm olarak kendini gösteren şiddetli bir formdur.
Karakteristik yüz özellikleri arasında büyük bir kafa, belirgin alın, düşük saç çizgisi, yuvarlak kaşlar, büyük kulaklar, burnun düzleştirilmiş köprüsü, çıkıntılı çene, geniş aralıklı dişler, büyümüş diş etleri ve büyük dil sayılabilir.
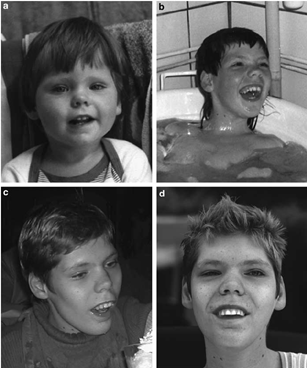
Şekil 1. Alfa-mannosidozda yüz özellikleri. A. 18 aylık ikizler. Genişlemiş baş, kısa boyun, yuvarlak kaşlar, yayılmış burun ve belirgin alın görülmektedir. B. Aynı ikizler 8 yaşında. Ellerin hafif kas atrofisi dikkat çeker. C. İşitme cihazı kullanan 27 yaşındaki alfa-mannosidoz hastası.
Diğer
belirtiler arasında şunlar olabilir:
Hareketleri
koordine etmede zorluk (ataksi)
Oturma
ve yürüme gibi motor becerilerinde gecikme
Konuşma
bozuklukları
İmmün
yetmezlik nedeniyle artan enfeksiyon riski
Karaciğer
ve dalağın genişlemesi (hepatosplenomegali)
Beyindeki
sıvı birikmesi (hidrosefali)
İşitme
kaybı
Göz
merceğinin bulanıklaşması (katarakt) ve yakını görememe gibi göz problemleri
Eklem
ağrısı ve iltihabı
Alfa-mannosidozu
olan bazı kişilerin depresyon, anksiyete veya halüsinasyonlar gibi zihinsel sorunları
vardır. Kalp ve böbrek problemleri de ortaya çıkabilir.
Prevalans
Alfa-mannosidozun
prevalansı hakkında çok az şey bilinmektedir. Avustralya’da yapılan bir
araştırmada, 500,000 kişide bir yaygınlık olduğu bildirilmiştir. Norveç’te yapılan
bir çalışmada 4.5 milyonluk bir nüfusta sadece altı kişide bu hastalığın
varlığı bildirilmiştir. Ayrıca Çekya’da 300,000 kişide bir kişi olarak
bildirilmiştir. Hastalık herhangi bir etnik gruba özgü değildir; dünyanın her
yerinden bireylerde tanımlanmıştır.
Alfa-mannosidozun
ayrıca dünya çapında yaklaşık 500,000 kişiden birinde meydana geldiği tahmin
edilmektedir.
Kalıtım Paterni
MAN2B1
genindeki mutasyonlar alfa-mannosidoza neden olur. Bu gen, alfa-mannosidaz
enzimini yapmak için talimatlar sağlar. Bu enzim lizozomlarda çalışır. Enzim
lizozomlar içinde, belirli proteinlere (glikoproteinler) bağlı oligosakkaritlerin
parçalanmasına yardımcı olur. Özellikle, alfa-mannosidaz enzimi mannoz adı
verilen şeker molekülü içeren oligosakkaritlerin parçalanmasına yardımcı olur.
MAN2B1 genindeki mutasyonlar, alfa-mannosidaz enziminin mannoz içeren oligosakkaritlerin parçalanmasındaki rolünü yerine getirme kabiliyetine müdahale eder. Bu oligosakkaritler lizozomlarda birikir ve hücrelerin arızalanmasına ve sonunda ölmesine neden olur.
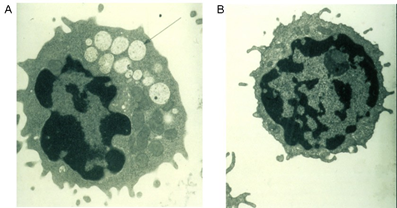
Şekil 2. Oligosakkarit birikimi sonucu koful oluşturan bir lenfosit ve normal lenfosit elektron mikrografisi. A. Alfa-mannosidoz hastasında koful oluşturan bir lenfosit. B. Normal olan bir lenfosit.
Dokular
ve organlar, oligosakkaritlerin anormal birikimi ve ortaya çıkan hücre ölümü
nedeniyle hasar görür ve alfa-mannosidozun karakteristik özelliklerine yol
açar.
Teşhis Yöntemleri Ve Tedaviler
Lizozomal
depo hastalığı olan alfa-mannosidozdaki ana klinik özellikler diğer lizozomal
depo hastalıkları ile örtüşebilir. Bununla birlikte, bu diğer lizozomal depo
hastalıkları ile ilişkili ayırt edici klinik özellikler, klinik
laboratuvarlarda biyokimyasal testlerin mevcudiyeti ve doğal geçmişlerinin
anlaşılması, bunların birbirinden ayırt edilmesine yardımcı olmaktadır.
Alfa-mannosidoz
ön tanısı konulan bir kişide hastalığın ve hastalık için gerekli kişisel
tedavilerin belirlenmesi için, aşağıdaki değerlendirmeler önerilir:
Hastanın
işitme kaybı, sinirlilik, depresyon; sosyal, ev, okul veya işle ilgili
aktivitelerde veya yürüme mesafesindeki değişiklik; ishal veya idrar tutamama,
kas ağrısı, eklem ağrısı, azalmış hareket aralığı ve kemik ağrısı gibi
şikayetlerinin tıbbi öyküsünün belirlenmesi gerekmektedir.
Otoskopi,
oftalmoskopi, karaciğer ve dalak boyutunun değerlendirilmesi, kalp ve
akciğerlerin oskültasyonu, yürüyüş dahil nörolojik durum ve eklem hareket
açıklığının ortopedik değerlendirmesini içeren fizik muayene yapılmalıdır. Çocuklarda
standart büyüme çizelgeleri kullanarak boy, ağırlık ve özellikle baş çevresinin
büyüklüğü ölçülüp büyüme değerlendirilmelidir.
İşitme
bozukluğu ve orta kulak enfeksiyonlarını tespit etmek için bir kulak burun
boğaz uzmanı tarafından muayene edilmelidir.
Kornea
opasiteleri, miyopi, hipermetropi ve şaşılık açısından oftalmolojik olarakdeğerlendirilmelidir.
Fonksiyonel
seviyeyi ve öğrenme kapasitesini belirlemek için nöropsikolojik testler
uygulanmalıdır.
Sistemik
lupus eritematozusu (SLE) hariç tutmak için klinik muayene ve immünolojik
testler (örn., Antinükleer antikorlar, anti-ds-DNA antikorları) ile kan
testleri yapılmalıdır.
Başın
düz radyografileri, dizler (ön-arka görünüm), omurga (yan görünüm) ve herhangi
bir semptomatik bölgenin iskelet değerlendirmesi yapılmalıdır.
Yaşlı
bireylerde osteopeni veya osteoporozu tespit etmek için kemik dansitometrisi
uygulanmalıdır.
Özellikle
hidrosefali belirtileri ve semptomları varsa (örneğin, baş ağrısı, artan
yürüyüş ataksisi, bulantı) ventriküllerin boyutunun ve serebellumun şeklinin ve
boyutunun değerlendirilmesi için beynin BT taraması yapılmalıdır.
Bir
klinik genetik uzmanı ve / veya genetik danışman ile konsültasyon.
Çok
semptomatik bir hastalığın karakteristik bulgularının tanımlanması üzerine
aşağıda açıklanan testlerin sonuçlarına dayanarak alfa-mannosidoz tanısı konulmasına
yardımcı olunabilir.
İdrarda
oligosakkaritler: İdrardaki mannoz açısından zengin oligosakkarit
konsantrasyonlarını ölçmek için bir ön araştırma yapılabilir. Mannoz zengini
oligosakkaritlerin yüksek idrar atılımı düşündürücüdür, ancak hastalığın tanısı
için yeterli değildir.
Alfa-mannosidaz
aktivitesi: Tanı, bir florometrik analiz yoluyla lökositlerde veya diğer
çekirdekli hücrelerdeki alfa-mannosidaz aktivitesinin ölçülmesi ile doğrulanır.
Bu test, genetik testle birlikte en güvenilir tanı yöntemidir.
Genetik
test: Hastalığa neden olan mutasyonun, periferik kan örneği kullanılarak polimeraz
zincir reaksiyonu (PCR) amplifikasyonu ile tanımlanması güvenilir bir tanı
yöntemidir.
Uygulanan Tedaviler
Konjenital
alfa-mannosidoz için bir tedavi yoktur ve genel olarak, ortaya çıkan
komplikasyonları önlemek amacıyla hastalığın yönetimi ele alınır. Aşılar,
antibiyotikler, işitme cihazları, gözlükler, ortopedik ve diğer yardımcı
cihazlar, eğitim müdahaleleri ve konuşma terapisi gibi bireysel semptomlara
yönelik tedaviler gerektiği şekilde önerilir. Sağlığı ve tedaviye yanıtı
izlemek için düzenli takip önerilmektedir.
Araştırılmakta Olan Tedaviler
Kemik iliği nakli
Alfa-mannosidoz
tedavisinde kemik iliği nakli denenmiştir. Kemik iliği nakli ile erken tanı ve
hızlı tedavi, bilişsel gerilemeyi önleme ve semptomları iyileştirme şansını
arttırır. Bununla birlikte, bu potansiyel tedavinin uzun vadeli güvenliğini ve
etkinliğini belirlemek için daha fazla araştırmaya ihtiyaç vardır. Prosedürü
ciddi komplikasyon riski taşır. Kemik iliği naklinden sonra, normal gelişim
sağlanamamasına rağmen, etkilenen bireyler gelişimsel ilerleme kaydetmiştir. En
iyi donör ailesel HLA-özdeş olandır, ancak çoğu zaman bu tip donör
tanımlanamaz, bu durumda enzim replasman tedavisi (ERT) en iyi seçenek olarak
belirlenebilir.
Gen Terapisi
Gen
terapisi ayrıca bazı lizozomal depo bozuklukları için olası bir tedavi olarak
araştırılmaktadır. Aktif enzim üretebilen normal genin kalıcı transferi göz
önüne alındığında, bu tedavi şeklinin teorik olarak bir tedaviye yol açması
muhtemeldir. Bununla birlikte, şu anda, gen terapisi başarılı olmadan önce
çözülmesi gereken birçok teknik zorluk vardır.
Allan – Herndon –
Dudley sendromu (AHDS) ciddi derecede zihinsel gelişim, dizartri, atetoid
hareketler, kas hipoplazisi ve spastik parapleji ile karakterize X’e bağlı bir
durumdur. Sadece erkeklerde meydana
gelen bu durum, gelişimi doğumdan önce bozar. Hastalık, etkilenen erkeklerde
konuşmayı ve iletişim yeteneğini kısıtlamış olsa da, diğer insanlarla iletişimde
eğleniyor gibi görünüyorlar.
Allan-Herndon-Dudley
sendromlu çocukların çoğunda zayıf kas tonusu (hipotoni) ve birçok kasın az
gelişmesi (kas hipoplazisi) durumu vardır. Yaşlandıkça, genellikle belirli
eklemlerin hareketini kısıtlayan kontraktür denilen eklem bozuklukları
geliştirir. Anormal kas sertliği (spastisite), kas zayıflığı ve kolların ve
bacakların istemsiz hareketleri de hareketliliği sınırlar. Sonuç olarak,
Allan-Herndon-Dudley sendromlu birçok insan bağımsız olarak yürüyemez ve yetişkinlikte
tekerlekli sandalye kullanımı mevcuttur.
Klinik Tanım
Hastalık
spastisiteye (kontraktürler, Babinski işareti ve klonus) ilerleyen ve
genellikle yaşamın erken döneminde saptanabilen konjenital hipotoni (doğumda
veya yaşamın ilk haftalarında / aylarında görülür) olarak kendini gösterir.
Hiperrefleksi, daha sonra yaşamda ortaya çıkar. Etkilenen erkekler de bebeklik
ve erken çocukluk döneminde, motor dönüm noktaları gecikmesi ve baş ve
desteklemede zorluk olarak ortaya çıkan kas hipoplazisi ve genel kas zayıflığı
ile kendini gösterir. Hastaların% 100’ünde hipotoni ve ciddi zihinsel eksiklik
vardır. Şiddetli psikomotor gecikme en başından itibaren mevcuttur (motor ve
dil dönüm noktalarının gecikmesi) ve özerkliğe asla ulaşılamaz. Yüz, zaman
içinde gelişen ayırt edici özelliklere sahiptir: açık ağız, çadırlı üst dudak, pitoz(sarkma)
, kulakların anormal katlanması, burun ve kulakların yumuşak dokusunun
kalınlaşması ve kalkık kulak memeleri. Uzun, ince eğik ayaklar da tipiktir.
Oküler belirtiler (yani döner nistagmus ve ayrık göz hareketleri) nadirdir.
Nöbetler ve kilo alım problemleri bazı hastalarda bildirilmiştir. Hipotoni ve
kas hipoplazisinin bir sonucu olarak düşünülen , pektus ekskavatum ve skolyoz
bulunabilir.
Epidemeyoloji
Bugüne kadar
literatürde 320 hastalıktan etkilenen bireye sahip en az 132 aile
bildirilmiştir. Yaygınlık bilinmemekle birlikte, bir çalışma, etiyolojisi
bilinmeyen zihinsel özürü olan erkeklerin% 1.4’ünde AHDS’yi tanımlamıştır.
Sadece erkekler etkilenir.
Kalıtım Kalıbı
Bu durum X’e bağlı resesif, kalıtsaldır. Hastalığa neden
olan mutasyona uğramış gen , iki cinsiyet kromozomundan biri olan X kromozomu
üzerinde bulunuyorsa, durum X’e bağlı olarak kabul edilir. Sadece bir X
kromozomu olan erkeklerde , her hücrede genin değiştirilmiş bir kopyasının
bulunması, hastalığa neden olmak için yeterlidir. İki X kromozomu olan
kadınlarda, bozukluğa neden olmak için genin her iki kopyasında da mutasyon
bulunmalıdır. Erkekler X’e bağlı resesif bozukluklardan kadınlardan daha sık
etkilenir. X’e bağlı kalıtımın bir özelliği, babaların X’e bağlı özellikleri,
oğullarına geçirememesidir.
Teşhis Yöntemleri
Tanı, klinik
bulgulara ve diğer tiroid hormonu serum düzeylerinin varlığına dayanır:
erkeklerde anormal derecede yüksek 3,3 ‘, 5’-triiyodotironin (T3), düşük ila
normal serbest tetraiyodotironin (T4) seviyeleri ve normal ila hafif yüksek
tiroid uyarıcı hormon (TSH) seviyeleri vardır. SLC16A2 genindeki mutasyonları
gösteren moleküler genetik test tanıyı doğrular.
Yönetim ve Tedavi
Şu anda, AHDS için
herhangi bir tedavi mevcut değildir ve yönetim destekleyici önlemlerden oluşur.
Fiziksel, mesleki ve konuşma terapisi faydalı olabilir. Distoni,
antikolinerjikler, L-DOPA, karbamazepin veya lioresol gibi bazı ilaçlarla
tedavi edilebilir. Nöbetler mevcut
olduğunda, standart antiepileptik ilaçlarla kontrol edilebilir. Hipotiroidizm
tedavisi yararlı görünmemektedir.
Hastalıkla İlgili Genler
AHDS, tiroid hormonu
T3 için spesifik bir taşıyıcı olan monokarboksilat taşıyıcı 8’i (MCT8) kodlayan
SLC16A2 genindeki (Xq13.2) mutasyonlardan
kaynaklanır . Nörolojik sorunlar, tiroid hormonu T3’ün bazı nöronal hücrelere
taşınamamasından kaynaklanabilir.
Prognoz
Birkaç hasta
60’larında hayatta kalmasına rağmen, çoğu hasta bağımsız olarak oturamadığı,
ayakta duramadığı veya yürüyemediği için genel yaşam beklentisi tehlikeye
giriyor ve yaşam kalitesi ciddi şekilde etkileniyor.





{kind=link}
{kind=link}