Tanım
Cornelia de Lange sendromu, vücudun birçok kısmını etkileyen gelişimsel bir bozukluktur. Bu bozukluğun özellikleri etkilenen bireyler arasında oldukça çeşitlilik gösterir ve görece hafif ya da şiddetli olabilir.
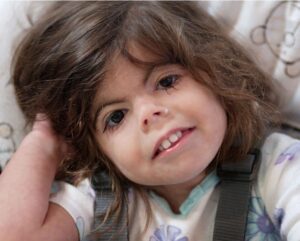
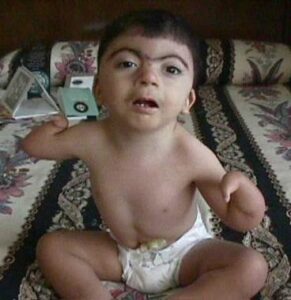
Hastalığın karakteristik özellikleri: doğum öncesi ve sonrası yavaş gelişim sonucu kısa boy, orta veya ileri derecede zeka geriliği ve kol, kafa ve parmak kemiklerindeki anormalliklerdir. Ayrıca Cornelia de Lange sendromu olan insanların çoğu; kemerli ve birleşik kaş, uzun kirpikler, aralıklı dişler, yukarı kalkık ve küçük burun ile kulakların başın aşağısında olması gibi belirgin yüz özelliklerine sahiptir. Bu hastalıktan etkilenmiş olan insanların birçoğunda, bir iletişim ve sosyal etkileşim hastalığı olan otizm benzeri davranışsal bozukluklar da görülmektedir.
Fazla miktarda vücut kılı, normalden küçük kafa, işitme kaybı ve sindirim sistemi problemleri de hastalığın belirti ve semptomlarından olabilir. Bu sağlık sorununa sahip insanların bazıları “yarık damak/yarık dudak” denilen ağız tabanında bir açıklıkla doğarlar. Nöbetler, kalp rahatsızlıkları ve göz sorunları da bu hastalığa sahip insanlarda gözlemlenmiştir.
Genetik Değişiklikler
Cornelia de Lange sendromunu, en az beş genin mutasyonu sonucu oluşabilir: NIPBL, SMC1A, HDAC8, RAD21 ve SMC3. NIPBL gen mutasyonu bu hastalığa sahip insanların yarısından fazlasında tespit edilmiş bulunmaktadır, diğer genler ise daha az yaygındır.
Bu beş genden üretilen proteinler, doğum öncesi gelişimde önemli rol oynayan “kohezin kompleksi”nin yapısına katılır veya işlevini yerine getirmesinde yardımcı olur. Kohezin kompleksi, hücre içinde kromozom yapısı ve organizasyonunun düzenlenmesine, hücrenin genetik bilgisini dengelenmesine ve zarar görmüş DNA’nın tamirine yardımcı olur. Bu kompleks aynı zamanda kol, bacak, yüz gibi vücut bölümlerinin gelişimine rehberlik eden genlerin aktivitesini düzenler.
NIPBL, SMC1A, HDAC8, RAD21 ve SMC3 genlerindeki mutasyonlar, kohezin kompleksinin faaliyetini zayıflatarak erken gelişimin önemli dönemlerinde gen regülasyonunu bozar ve Cornelia de Lange sendromuna sebep olurlar.
Cornelia de Lange sendromunun özellikleri oldukça çeşitlilik göstermekte ve aynı gen mutasyonuna sahip bireylerde bile hastalığın şiddeti farklılık gösterebilmektedir. Bilim insanları, fazladan genetik veya çevresel faktörlerin bireye özgü belirti ve semptomların belirlenmesinde önemli olduğunu düşünmektedirler. Genel olarak, SMC1A, RAD21 ve SMC3 gen mutasyonları, NIPBL geni mutasyonuna göre daha hafif şiddette belirti ve semptoma sebep olmaktadır. HDAC8 genindeki mutasyonları ise biraz daha farklı sonuçlar vermektedir. Örneğin: bebeklerde kafadaki yumuşak dokunun geç kapanması (bıngıldak), ayrık gözler ve dental anormallikler. NIBPL gen mutasyonuna sahip bireyler gibi, HDAC8 gen mutasyonuna sahip bireylerde ileri dercede zeka geriliğine sahip olabilirler.
Cornelia de Lange sendromu vakalarının yaklaşık yüzde otuzunun sebebi bilinmemektedir. Bilim insanları bilinen beş gendeki fazladan değişikliklerin ve başka genlerdeki mutasyonların bu hastalığa sebep olabileceğini düşünmektediler.
Kalıtım
Cornelia de Lange sendromu NIPBL, RAD21 veya SMC3 genlerindeki mutasyonlar sonucu oluşsa da, otozomal baskın bir şekilde kalıtıldığı düşünülmektedir. Otozomal baskın kalıtım, hücrelerde bir değişmiş kromozomun olmasının hastalığın ortaya çıkması için yeterli olması demektir. Vakaların çoğu, yeni gen mutasyonlarının sonucu ve hastalık aile geçmişinde bulunmayan kimselerde görülmektedir.
Cornelia de Lange sendromu HDAC8 veya SMC1A genlerinde ortaya çıkan mutasyonla oluştuğunda hastalık X kromozomuna bağlı olarak baskın bir şekilde kalıtılır. Mutasyona uğrayan gen X kromozomu (cinsiyet kromozomlarından biri) üzerinde bulunuyorsa hastalık X-bağımlı olarak kabul edilir. X-bağımlı Cornelia de Lange hastalığı hakkında yapılan araştırmalar, hücrede, değişime uğrayan genin bir adedinin bulunmasının hastalığa sebep olmak için yeterli olduğunu göstermektedir. Erkeklerin kadınlara göre daha sık veya daha şiddetli etkilendiği X-bağımlı çekinik kalıtımın aksine, X-bağımlı baskın kalıtımda erkekler ve kadınlar benzer şekilde etkilenir. Birçok vaka HDAC8 veya SMC1A genlerindeki yeni mutasyonlar sonucu oluşur ve hastanın aile geçmişinde bu hastalık bulunmaz.
Tanı
Moleküler testler ilgili genlerde bir mutasyon tespit etmişse birey CdLS sahibidir. Aksi halde, klinik sonuçları, yüz kriterlerine uymalı, bu kriterler, altı sistem kategorisinden en az iki veya üçüne ait olmalıdır. Sistemlerden en az biri büyüme, gelişme veya davranışla ilgili olmalıdır. Bu kriterlere uyuyorsa bireye klinik olarak CdLS teşhisi konmuştur.
Tanı Koyma
- CdLS gen testinde pozitif muyaston VEYA
- Yüzsel bulgular ve ana kategorilerden birine giren iki kriter VEYA
- Yüzsel bulgular ve ana kategoriye giren bir, fazladan da iki kriter (ana veya yan kategoriye ait)
Yüzsel Bulgular
- Birleşik kaş VE >aşağıdakilerden üç veya daha fazlası
- Uzun kirpikler
- Kısa burun, öne eğik burun delikleri
- Üst dudak ve burun arasında uzun ve belirgin alan
- Geniş ya da basık burun kemeri
- Küçük ya da kare çene
- İnce, köşeleri aşağıya dönük dudak
- Yüksek damak
- Geniş aralıklı ya da eksik diş
Ana Kategori
BÜYÜME >aşağıdakilerden iki veya daha fazlası
- Yaşıtlarından yüzde beş daha az ağırlık
- Yaşıtlarından yüzde beş daha az boy
- Yaşıtlarından yüzde beş daha küçük ölçekte fetüs kafası
GELİŞME >aşağıdakilerden bir veya daha fazlası
- Gelişim geriliği veya zeka geriliği, (kas yeteneğindense konuşma becerisi daha çok etkilenmiştir)
- Öğrenme zorlukları
DAVRANIŞ >aşağıdakilerden iki veya daha fazlası
- Dikkat dağınıklığı ve hiperaktivite
- Obsesif-kompülsif davranışlar
- Anksiyete
- Sürekli dolaşma ihtiyacı
- Sinirlilik
- Kendine zarar verme
- Aşırı utangaçlık ve çekingenlik
- Otizm benzeri hareketler
Yan Kategori
KAS VE İSKELET İLE İLGİLİ >aşağıdakilerden bir veya daha fazlası
- Eksik kol veya önkol
- Küçük el ve/veya ayaklar, el ve ayakta eksik parçalar
- Kıvrılmış serçe parmak
- Avuç içinde anormal katlanmalar
- Çıkık/anormal dirsek
- Kısa başparmak eklemi
- El kemiğinin başparmakla birleştiği yerde şekil bozukluğu
- Ve 3. Ayak parmakları arasında kısmi ağ
- Omurga eğriliği
- Göğüs veya sternum deformasyonu
- Çıkık kalça veya displazi
SİNİRSEL/DERİ >aşağıdakilerden üç veya daha fazlası
- Düşük göz kapağı
- Gözyaşı kanalı bozukluğu ya da gözkapağı iltihabı
- Miyop
- Ciddi göz şekil bozukluğu veya retina incelmesi
- Sağırlık ya da işitme kaybı
- Nöbetler
- Alacalı deri
- Fazla miktarda vücut tüyü
- Küçük göğüs ucu ve/veya göbek deliği
DİĞER ÖNEMLİ SİSTEMLER >aşağıdakilerden üç veya daha fazlası
- Sindirim sistemi şekil/konum bozuklukları
- Diyafragmatik fıtık
- Mide ve yemek borusunda reflü
- Yarık damak
- Konjenital kalp rahatsızlıkları
- Mikropenis
- Anormal yerleşmiş üretra açıklığı
- Börek/idrar torbası şekil bozuklukları
Tedavi-Kontrol
Cornelia de Lanfe sendromuna sahip olan hastalara yönelik ilgi, yaş ile bağlantılıdır. Bebek ilk, çocukluk, gençlik ve yetişkinlik çağındaki hastaların ihtiyaçları farklı olur.
Bebeklikte ve Tanı Koyulma Zamanında
Potansiyel CdLS tanısı düşünülüyorsa karyotip elde edilmelidir. (Kandaki kromozomlar değerlendirilmelidir). Tanı koyulduktan sonra önerilen hizmetler aşağıdaki gibidir:
- Ekokardiyogram
- Renal ultrason
- Pediyatrik göz muayenesi
- İşitme değerlendirmesi (otoakustik emisyon veya beiyin sapının uyarılması ölçülür).
- UGI testi (sindirim sisteminin üst kısımlarının – yemek borusu, mide, duodenum- X-Ray ile taranması. Malrotasyon ya da reflü var mı diye bakılır).
- Gastraözofogal reflü taraması (ph ölçümü ve/veya endoskopi yapılır. Varsa ilaç veya ameliyatla tedavi edilir)
- Gelişme değerlendirmesi (bebeklikte ve sonrasında her 1-3 yılda bir)
- Erken müdahale hizmetleri başlar ve ihtiyaçlar karşılana kadar devam eder
- Uygun CdLS gelişim tablosu takip edilerek gelişme değerlendirmesi yapılır (genellikle yüksek kalorili besinler önerilir fakat bireyler hızlı metabolizmayla kendi tempolarında büyümeye devam ederler)
- Destek veren kurumların iletişim bilgileri ailelere verilir
- İleriki hamileliklere yönelik moleküler test önerilir
Erken Çocukluk(Bir İla Sekiz Yaş Arası)
Birey düzenli değerlendirme ve aşılamaya tabi tutulmalıdır.
- Erkeklerde yerine inmemiş testisler 18 aya kadar düzetilmelidir
- Devam eden gelişim hizmetleri, okul, terapi gibi meseleler bireyselleştirilmelidir. Bireyler fiziksel, mesleki ve konuşma terapilerinden faydalanacaktır. İşaret dili öğrenimi, sözlü iletişimi kolaylaştıracağından, tavsiye edilmektedir.
- CdLS gelişim tablolarından bireyin büyüme/gelişmesi takip edilmeye devam edilir.
- Altı ayda bir diş muayenesi
- İlk değerlendirmenin sonuçlarına göre bir kere veya yıllık pediyatrik göz değerlendirmesi
- Her iki-üç yılda bir işitme testi
- Sindirim sistemi rahatsızlıkları, reflü ihtimaline karşı sürekli olarak değerlendirilmelidir. Endoskopi en büyük verimi verse de ph ölçeği de düşünülmelidir.
- Bağırsak düğümlenmesinin herhangi bir belirtisine karşın direk acile getirilmelidir, ameliyat gerekebilir.
- Operasyon gerçekleşirse, sonrasında uzmanlar tarafından takibe alınmalıdır
- Herhangi bir ameliyat gerçekleştiğinde, anesteziden daha çok verim alabilmek amacıyla, alakalı uzmanların tamamının görüşleri dikkate alınmalıdır. Böylelikle aynı zamanda birey, ihtiyaç duyduğu şekilde tanısal veya kontrol amaçlı incelenmiş olur.
- Bebekliğin j,k maddeleri.
Geç Çocukluk (Sekiz Yaşından Ergenliğe Kadar)
Birey düzenli değerlendirmeye tabi tutulmalıdır.
- Eklem kasılmaları, kalça sorunları, nasır, omurga eğriliği ve ortotik şikayetler için ortopedist ile görüşümelidir.
- ADHD,kendine zarar verecek şekilde davranışlar gibi sorunlar baş gösterirse davranışsal değerlendirmeye tabi tutulmalıdır.
- Erken çocukluğun b,k maddeleri.
Gençlik (Ergenlikten 20 Yaşına Kadar)
Birey düzenli değerlendirmeye tabi tutulmalıdır.
- Devam eden gelişim hizmetleri. Okul yerleştirilmesi ve terapi bireyselleştirilmelidir. Okul veya liseden sonra işe yerleştirme, mesleki eğitim ve/veya yükseköğrenim için erkenden plan yapılmalıdır.
- Kadınlar için geç ergenlikten başlayarak yetişkinlik süresince, cinsel aktiviteye bağlı olarak, en az üç yılda bir Pap Smear testi ile pelvik muayene düşünülmelidir. Hasta ve ailesiyle adet döngüsü düzeni ve doğum kontrol ile ilgili hormon tedavisi hakkında görüşülmelidir. (hasta ve ailesine göre spesifik)
- Gelişimsel olarak yatkınlığı varsa nüks riski hakkında aileye konuşulmalıdır.
- Geç çocukluğun a,c maddeleri.
Yetişkinlik
Birey düzenli değerlendirmeye tabi tutulmalıdır.
- Kan basıncı takip edilmeli, baseline EKG düşünülmeli, düzenli göğüs, testis ve prostat muayenesi olunmalıdır.
- Daha yüksek eğitim, iş meseleleri veya mesleki hazırlık konuşulmalıdır.
- ADHD, OCD, kendine zarar verecek davranışlar ve depresyon gibi durumlar ortaya çıkarsa davranışsal veya psikiyatrik değerlendirme yapılmalıdır
- Osteoporoz oluşup oluşmadığını anlamak için DEXA scan kullanılmalıdır
- Şikayetlere göre her 4-6 ayda bir, özel ihtiyaçları olan ailelere aşina dişçiler tarafından diş muayenesi olunmalıdır
- Gençliğin b,d maddeleri.
Sendromun Diğer İsimleri
- BDLS
- Bracgmann de Lange Sendromu
- CdLS
- de Lange sendromu
- typus degenerativus amstelodamensis


Kaynakça
1) https://www.omim.org/
2) https://rarediseases.info.nih.gov/diseases
3) https://www.orpha.net/consor/cgi-bin/Disease_Search.php
4) https://ghr.nlm.nih.gov/condition
5)https://rarediseases.org/for-patients-and-families/information-resources/rare-disease-information/