Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Demodikozis, başta yüz bölgesi olmak üzere derinin belirli bölgelerini etkileyen nadir bir enfeksiyon hastalığıdır. Demodeks akarları olarak bilinen bir tür iç paraziti bu hastlığa sebep olmaktadır. Demodex folliculorum ve Demodex brevis olmak üzere vücutta iki türü bulunan bu parazitler herhangi bir genetik değişikliğe sebep olmamakla birlikte, hemen hemen her yaş grubundaki insanların cilt florasında bulunmaktadır. Özellikle, yanak, alın, burun kenarları ve kulak çevresi gibi yüz bölgelerinde yaygındırlar. Genelde zararsız durumdadırlar. Ancak, sayıları arttığı takdirde demodikozis adı verilen cilt rahatsızlığına sebep olmaktadır. Normalde yoğunluğu az olan Demodex parazitleri, sayıları arttığında bağışıklık sistemini bir parçası olan toll-benzeri reseptörleri (TLR) üzerinden etkileşime geçerek deride enfeksiyon belirtilerine sebep olduğu düşünülmektedir. Bağışıklık sistemi üzerinden etkileşime geçen bu parazit türü genelde; bağışıklık sisteminin düşük olması, bağışıklık sistemini baskılayan ilaçların kullanımı, kemoterapi tedavisi almak ve AIDS gibi bağışıklığın baskılanması durumlarında daha yüksek ihtimalle görülebilir.
Belirti ve Semptomlar
Demodikozis hastalığının en yaygın ve ilk belirtileri yüzde görünür. Ve şu belirtilere sebep olmaktadır;
Yanma hissi
Kaşıntı
Pürüzlü yüzey
Kızarık hassas cilt
Beyaz folikülümsü görüntü
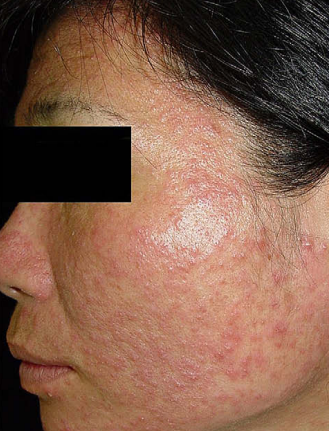
Figur 1: Demodikozis hastalığına sahip 44 yaşındaki bir kadın hastanın cildinin görüntüsü. [6]
Aynı zamanda gözleri de etkileyen bu hastalık ise gözlerde şu belirtilere sebep olmaktadır;
Gözde kaşıntı
Gözde tahriş
Görme duyusunda azalma
Kirpik dökülmesi
Pullu ve beyaz kalıntılar olan göz kapağı görüntüsü
Görülme Sıklığı
Demodikozis hastalağının speesifik bir görülme sıklığı olmadığı pek çok çalışma ile kanıtlanmıştır. Kadın ve erkek hastalar arasındaki kıyaslamaya bakıldığında da anlamlı bir ayrım bulunamamıştır. Ancak, yapılan bir araştırmada 16-96 yaş grubunda görülme sıklığı belirli aralıklarla değişse de genel oran %70’den fazla olarak belirtilmiştir.
Kalıtım Paterni/Deseni
Kalıtsal olmayan demodikozis hastalığı, parazit kaynaklı olmasından dolayı genetik yollarla aktarılamaz. Bu sebeple 15 yaş ve altı, özellikle yenidoğanlarda, bu parazite genelde rastlanmaz veya düşük orandadır. Ancak, zamanla insanlarla temastan dolayı ileri yaşta bu parazitlere hemen hemen herkeste rastlanmaktadır.
Teşhis Yöntemleri ve Tedaviler
Teşhis yöntemlerinin başında ise belirtilerin değerlendirilmesi sonrasında, dermateskop ile bölgeyi inceleme yöntemi gelmektedir. Dermateskop sayesinde parazitlere ait kalıntılar gözlemlenebilmektedir. Onun haricinde benzer bir yöntem olan, hastanın derisinden sürüntü örneği alma ve mikroskop altında analiz etme yöntemi de kullanılmaktadır.
Tedavi olarak ilaç tedavileri ve deri üstünden kimyasal kullanımı olmak üzere bir kaç çeşitli tedavi yöntemi bulunmaktadır. En yaygın olanı ise antibiyotik olan metronidazol kullanımıdır. Benzen heksaklorür olarak bilinen Lindan kullanımı da yaygındır. Ayrıca, bazı araştırmalarda da salisilik asit, selenyum sülfat ve potasyum hidroksit de tedavi amaçlı kullanılmış ve sonuç almıştır.
Hastalığın Diğer İsimleri
Demodikidozis
Demodikozis
Demodeks
Referanslar
Yazısız, H., Çekin, Y., & Koçlar, F. G. (2019). The Presence of Demodex Mites in Patients with Dermatologic Symptoms of the Face. Turkiye parazitolojii dergisi, 43(3), 143–148. https://doi.org/10.4274/tpd.galenos.2019.6062
Hsu, C.-K., Hsu, M. M.-L., & Lee, J. Y.-Y. (2009). Demodicosis: A clinicopathological study. Journal of the American Academy of Dermatology, 60(3), 453–462. doi:10.1016/j.jaad.2008.10.058
HUGUL, H., KECİCİ, A. S., & KUTLUBAY, Z. (2022). Treatment of Demodicosis with Potassium Hydroxide Dermabrasion. Practical Dermatology.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Ekstremite Kuşağı Kas Distrofisi hastası insanların vücut kasları genel olarak hasta olmayan insanlara kıyasla daha zayıftır. Özellikle üst bacak kasları, el ve kol kasları bu hastalıktan büyük oranla etkilenir ve hastalar kolayca yürüyemez ve gündelik işlerini yerine kolayca getiremez. [1] Konu başlığı olan otozoman dominant ekstremite kuşağı musküler distropi 1c hastalığı ise normal ekstremite kuşağı kas distrofisi hastalığının otozomal dominant halidir. Hastalarda Cav-3 geni tarafından üretilen caveolin-3 proteini eksiktir. Bu hastalığın etkisi yaş ilerledikçe güçlenmektedir. Kalp kası ve nefes almakta kullanılan kaslar genel olarak bu nadir hastalıktan etkilenmedikleri için bu nadir hastalığa sahip insanların ömür uzunlukları hasta olmayanlardan farklı değildir. [1]
Belirti ve Semptomlar
Genel olarak kaslarda zayıflık. Tırmanma, yürüme, ağır kaldırma gibi gündelik, kas gücü gerektiren işlerde zorluk çekmek. [1]
Genetik Görülme Sıklığı
1-6 / 100.000 [2]
Kalıtım Paterni/Deseni
Otozomal Dominant
Teşhis Yöntemleri ve Tedaviler
Kas biyopsisi, Serum kreatin kinaz kan testi, Kan üzerinden DNA testi ile Cav 3 genindeki mutasyonların saptanması. Otozomal Dominant Ekstremite Kuşağı Musküler Distropi 1C hastalığının şu ana kadar bilinen bir tedavisi yoktur. Ancak semptomlarla karşılaşan insanların yorucu işlerden uzak durması, düzenli ve yeterli olarak uyuması/dinlenmesi, sigara içilmemesi gibi yöntemlerle hastalığın etkilerini azaltabilmektedirler.
Chu, M. L., & Moran, E. (2018). The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon?. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics, 15(4), 849–862. https://doi.org/10.1007/s13311-018-0648-x
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Vasküler Ehlers-Danlos Sendromu genlerle aktarılabilen bir bağ doku hastalığıdır ve kusurlu kolajen proteinleri bu hastalığa sebep olur. Diğer Ehlers-Danlos Sendromları (EDS) arasında en zorlusu olarak bilinir. COL3A1 genindeki bir mutasyondan kaynaklanan bu hastalık nadiren COL1A1 geninki bir mutasyondan da ortaya çıkabilir. Belirtileri arasında ince, yarı saydam ve kolay morarabilen bir cilt, karakteristik bir yüz görünümü ve kırılgan damarlar, iç organlar gösterilebilir. [1]
Belirti ve Semptomlar
Anormal şekilde sık görülen kanamalar, kirpiklerde anomali, kalp kapakçığınde anomali, anormal yüz görüntüsü, yarı-saydam ve ince deri, kolay morarma ve yaşlı görünme en sık görülen semptomlardır. Nadiren görülen semptomlar arasında vertigo, geçiçi iskemik atakları ve uyku apnesi görülebilir. [1]
Genetik Görülme Sıklığı
Ehlers-Danlos Sendromu yalnızca beş bin kişiden birinde görülmekte iken daha nadir olan Vasküler Ehlers-Danlos Sendromu yalnızca iki yüz – iki yüz elli bin kişide bir görülmektedir. [2]
Kalıtım Paterni/Deseni
Vasküler Ehlers-Danlos Sendromu otozomal dominant olarak sonraki kuşaklara aktarılır. [1]
Teşhis Yöntemleri ve Tedaviler
Teşhis yöntemi olarak doktorlar hastalarının derilerini görsel olarak inceler, baskı uygular ve acı testleri yapar. Hastanın yüz şekli dikkate alınır. Hastanın geçmiş hastalıkları araştırılır. Kan testi uygulanır. [1]
Vasküler Ehlers-Danlos Sendromunun bilinen bir tedavisi mevcut değildir. Doktorlar yalnızca semptomları azaltma konusunda yardımcı olabilmektedirler.
Hastalıkla İlişkili Genler
Vasküler Ehlers-Danlos Sendromunun COL3A1 ve COL1A1 genlerinin düzgün çalışmaması ile ortaya çıktığı bilinmektedir. [1]
U.S. Department of Health and Human Services. (2021). Vascular Ehlers-Danlos syndrome – about the disease. Genetic and Rare Diseases Information Center. Retrieved December 21, 2022, from https://rarediseases.info.nih.gov/diseases/2082/vascular-ehlers-danlos-syndrome
Vascular Ehlers-Danlos Syndrome: Causes, symptoms and treatment. Cleveland Clinic. (2022). Retrieved December 21, 2022, from https://my.clevelandclinic.org/health/diseases/22696-vascular-ehlers-danlos-syndrome#management-and-treatment
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Epidermolizis Bulloza Simpleks, dermo- epidermal bileşkenin bazal membranı üzerinde kabarcık görünümünün meydana gelmesi ile karakterize edilen oluşumunda genetik yapının etkili olduğu deri hastalıklarındandır. Hastalığın on yedi alt tipi bulunmaktadır. Alt tiplerinin çoğu, bazal hücre keratinlerinin genlerindeki mutasyonun yanı sıra plektin, a6b4 intergrin, desmoplakin, plakoglobin ve plakophilin-1 genlerindeki mutasyondan kaynaklanmaktadır [1].
Plektin geninin kaybı veya belirgin şekilde azalması Kas Distrofisi İle Epidermolizis Bulloza Simpleks’e yol açmaktadır. Bu hastalıkla mücadele eden kişilerde, doğumda veya doğumdan kısa bir süre sonra cilt ve mukoza zarlarında kabarcıklar gözlenmektedir. Bu cilt belirtileri mine hipoplazisi (az gelişimi) ve tırnak distrofisi ile ilişkilidir. Karakteristik olarak, ilerleyici Kas Distrofisi yaşamın ilerleyen dönemlerinde ortaya çıkmaktadır. Fakat, belirtilerden biri olan kas tutulumunun başlangıcı değişkendir. Bazı durumlarda, erken bebeklik döneminde fark edilirken, bazı durumlarda ise 20 veya 30 yaşına kadar fark edilememektedir. Genellikle, kas tutulumu zayıflıkla sonuçlanmaktadır ve bu durum kişiyi tekerlekli sandalyeye mahkum hale getirmektedir [2].
Belirti ve Semptomlar
A. Alt ekstremitelerde kas distrofisi belirgindir. Küçük sürtünmelerden sonra genelleştirilmiş kabarcıklar [2]
B. Skarlı alopesi (saç köklerinin zarar görerek yerine skar dokusunun (fibröz yara dokusu) geçtiği kalıcı saç dökülmesi) [2]
C. Tırnaklarda şekil bozukluğu [2]
D. Deride genelleşmiş kabarma ve erozyonlar [2]
E. Torasik (sırt, göğüs) ve lomber (bel) omurganın şiddetli skolyozu [1]
F. Küçük sürtünme sonucu oluşan kabarcıklar [2]
G. Kas atrofisi (Çalışmayan kasın hacminin ve gücünün azalmasıdır. Kasın körelmesidir.) [2]
H. El ve ayaklardaki eklemler üzerindeki cilt lezyonları (epidermolizis bulloza) [3]
Göz
Pitoz (düşük göz kapağı)
Kısıtlı göz hareketleri
Göz çevresinde kabarcıklar
Ağız
Oral mukozal kabarcıklar
Diş çürükleri
Solunum
Boğuk ağlama, öksürük veya diğer solunum problemleri
Cilt, Tırnaklar, Saç
Palmoplantar keratoz (el içi ve ayak tabanı derisinin aşırı kalınlaşması)
Özellikle ayaklarda olmak üzere hafif yaralanma veya sıcaklık değişikliği sonucu ciltte meyada gelen kabarcıklar
Kalp, beyin, gastrointestinal, kemik veya böbrek sorunları
Ses
Kısık ses
Kas ve yumuşak dokular
Artan bağ dokusu
Dejeneratif değişiklikler
Miyofibriler düzensizlik
Belirgin şekilde değişen lif çaparı
Progresif kas distrofisi [5, 6]
Genetik Görülme Sıklığı
Prevelansı <1/1.000.000 şeklindedir [4].
Kalıtım Paterni/Deseni
EBS-MD, progresif kas distrofisi ve kabarcıklı cilt değişiklikleri ile karakterize edilen otozomal resesif bir hastalıktır. Erken çocuklukta ortaya çıkmaktadır [6]. Fakat bazı hastalarda bu durum farklılık göstermesi sebebiyle çocukluk veya yetişkinliğin sonlarına kadar ortaya çıkmayabilir. Bu hastalığın ölümcül bir sonucu olabilmektedir [4]
Teşhis Yöntemleri ve Tedaviler
Teşhis Yöntemleri
Cilt biyopsisi
Deri örneklerinin mikroskop altında özel testleri
Kansızlık için kan testi
Bakteriyel enfeksiyonu kontrol etmek için kültür
Diğer organlara bakmak için görüntüleme çalışmaları [5].
Doğum öncesi tanı, patojenik varyantın önceden teşhis edilmiş olduğu ailelere prenatal tanı önerilmelidir.
Genetik danışmanlık
Tedavi
Spesifik bir tedavisi bulunmamaktadır. Tedavi yara yönetimi ve genel destek ile semptomatiktir [4].
Hastalıkla İlişkili Genler
[9]
EBS-MD, 8q24 kromozomu üzerindeki insan plektin geninin (PLEC1) mutasyonlarının sebep olduğu nadir bir EBS varyantıdır. Plektin, çeşitli hücre tiplerinde ifade edilen, yüksek moleküler ağırlığa sahip çok yönlü bir hücre iskeleti bağlayıcı proteinidir [1]. PLEC geni, plektin adı verilen bir proteinin yapılması için talimatlar sağlamaktadır. Bu protein, cilt ve kas dahil olmak üzere vücutta bulunan çeşitli birçok dokuda üretilmektedir. Plektin, hücrenin yapısal çevresini (hücre iskeleti) oluşturan birkaç molekülle etkileşime girmektedir.
Genin diğer isimleri:
EBS1
EBSO
HD1
Hemidesmozomal protein 1
PCN
PLEC1
PLEC1_HUMAN
plectin1
PLTN [7]
Hastalığın Diğer İsimleri
Kas distrofisi olan EBS
Epidermolizis bulloza simpleks ile ekstremite kuşağı kas distrofisi
EBS-MD
MDEBS
MD-EBS [4,8]
Kaynaklar
[1] Kyrova, J., Kopeckova, L., Buckova, H., Mrazova, L., Vesely, K., Hermanova, M., Oslejskova, H., & Fajkusova, L. (2016). Epidermolysis bullosa simplex with muscular dystrophy. Review of the literature and a case report. Journal of dermatological case reports, 10(3), 39–48. https://doi.org/10.3315/jdcr.2016.1231
[2] Shimizu, H., Takizawa, Y., Pulkkinen, L., Murata, S., Kawai, M., Hachisuka, H., … & Nishikawa, T. (1999). Epidermolysis bullosa simplex associated with muscular dystrophy: phenotype-genotype correlations and review of the literature. Journal of the American Academy of Dermatology, 41(6), 950-956.
[9] Liu, C. G., Maercker, C., Castañon, M. J., Hauptmann, R., & Wiche, G. (1996). Human plectin: organization of the gene, sequence analysis, and chromosome localization (8q24). Proceedings of the National Academy of Sciences of the United States of America, 93(9), 4278–4283. https://doi.org/10.1073/pnas.93.9.4278
Cri-du-chat sendromu doğuştan itibaren hastanın büyüme ve gelişmesini olumsuz yönde etkileyen bir rahatsızlıktır. Bu rahatsızlığa sahip olan yenidoğanlarda sıklıkla tiz bir kedi benzeri ağlama, mikrosefali ve karakteristik yüz özellikleri (hipertelorizm, düşük kulaklar, küçük bir çene ve yuvarlak bir yüz gibi) görülür. Solunumda zorlanma ve beslenmede zorluk görülebilir. Bu hastalığa sahip bireylerde zihinsel bir engelin yanında gelişimde ve konuşmada gecikme görülür, ayrıca davranış bozuklukları da görülebilmektedir. Bu sendrom kromozom 5’in p kolu denilen bölgesindeki bir eksikliğe bağlı olarak görülmektedir. Genellikle semptomların şiddeti kromozom 5’te bulunan eksikliğin bölgesine ve büyüklüğüne bağlıdır. Bu eksiklik embriyonun erken aşamalarında meydana gelir ve genellikle ailesel bir kalıtıma bağlı değildir.
Semptomlar
1- Anormal ses
Çok sık görülmektedir.
2- Kedi-benzeri ağlama
Hastalığa ismini veren bu semptom oldukça tiz bir kedi benzeri ağlama ile karakterizedir.
Çok sık görülmektedir.
3- Epikantus
Epikantus ya da epikantal kıvrım, göz çevresinde bulunan kıvrılmış deri parçasıdır. Kıvrımın kirpikleri içe döndürüp göze zarar vermeye başladığı durumlarda tedavi edilir. Tedavi edilmemesi durumda başka hastalıklara sebebiyet verebilir.
Çok sık görülmektedir.
4- Ciddi seviyede zihinsel gerilik
IQ seviyesi 20 ila 34 arasında değişmektedir.
Çok sık görülmektedir.
5- Mikrosefali
Baş çevresinin yaş ve cinsiyete göre ortalamanın altındaki iki standart sapmadan daha küçük olduğu nörogelişimsel bir bozukluktur.
Çok sık görülmektedir.
6- Mikroretrognati
Alt çenenin geride olması durumu.
Çok sık görülmektedir.
7- Yuvarlak yüz
Çok sık görülmektedir.
8- Ciddi düzeyde gelişim eksikliği
Çok sık görülmektedir.
9- Aşağı eğimli palpebral fissürler
Gözlerin dış köşelerinin aşağıya doğru meyilli olduğu durum.
Sıklıkla görülmektedir.
10- Hipertelorizm
Gözler arasındaki mesafenin abartılı bir seviyede artması.
Sıklıkla görülmektedir.
11- İntrauterin gelişme geriliği
Anne rahmindeki bebeğin, gebelik haftasına göre olması gerekenden küçük olmasıdır.
Sıklıkla görülmektedir.
12- Skolyoz
Spinal deformiteler içerisinde en sık karşılaşılan ve ilerleyen evrelerde son derece ciddi duruş bozukluklarına neden olan üç boyutlu bir omurga deformitesidir. Bu deformite omurganın yapısal bozukluğundan kaynaklı olarak ortaya çıkabileceği gibi omurga dışı sebeplere bağlı olarak da gelişebilir.
Sıklıkla görülmektedir.
13- Kısa boyun
Sıklıkla görülmektedir.
14- Küçük el
Sıklıkla görülmektedir.
15- Kardiyovasküler sistem morfolojisinde anormallikler
Kalp ve damarlarda yapısal bozukluklar.
Zaman zaman görülmektedir.
16- Kasık fıtığı
Karın içindeki organların (ince bağırsaklar, bağırsak yağları gibi), karın duvarındaki zayıf bölgelerden çıkarak cilt altında şişlik oluşturmasıdır.
Zaman zaman görülmektedir.
17- Eklem hipermobilitesi
Eklemlerin normalden çok daha fazla hareket aralığının bulunması durumu. Bu rahatsızlığa sahip kişiler eklemlerinin fazla hareketine bağlı ağrı çekebilirler.
Zaman zaman görülmektedir.
Görülme Sıklığı
Prevalansının 15.000’ de 1 ila 50.000’de 1 olduğu düşünülmektedir. Toplumda tanısı konulmamış hastaların varlığı sebebiyle bu aralık konusunda kesinlik vermek zordur. Görülme sıklığındaki başka bir önemli detay ise kadın hastaların oranı erkek hastalardan daha yüksektir.
Genetik Paterni/Deseni
Çoğu vakada ailesel bir kalıtım görülmez. Eksiklik, ya da silinme, çoğu ya üreme hücrelerinin(sperm ya da yumurta) gelişimi sırasında ya da embriyonun erken gelişimi sırasında rastgele bir şekilde meydana gelir.
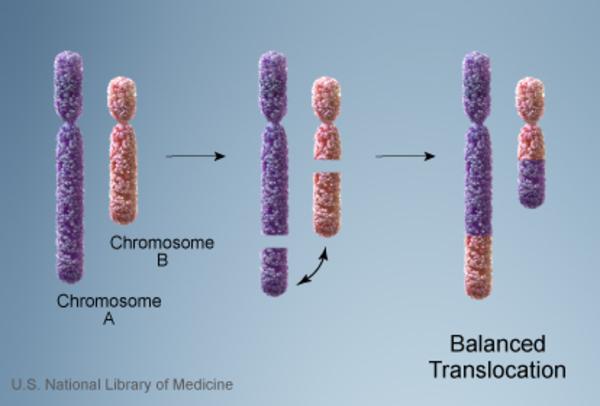
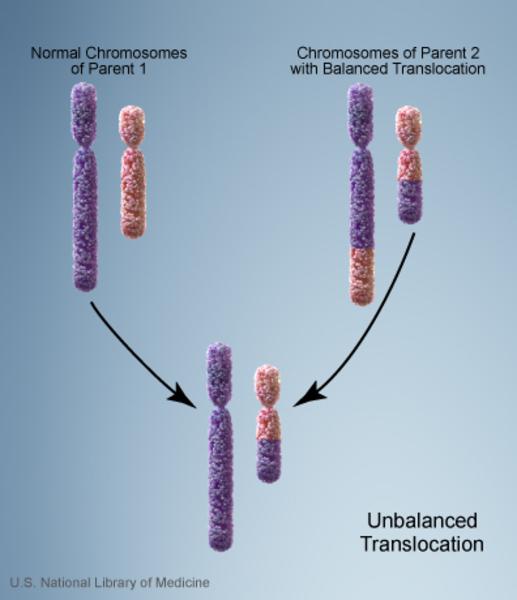
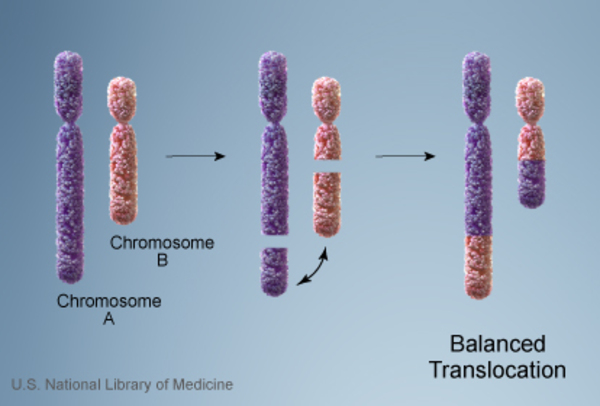
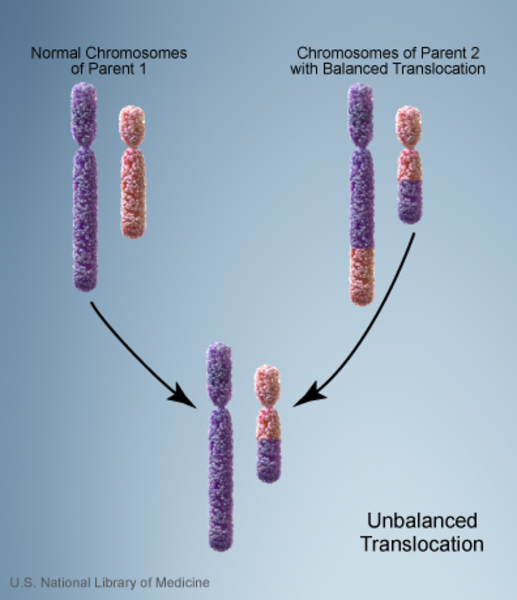
Vakaların %10’unda kromozom anormalliği hasta olmayan bir ebeveynden gelmektedir. Bu tarz durumlarda ebeveyn dengeli translokasyon adı verilen bir kromozomal yeniden düzenlemeye sahiptir. Dengeli translokasyonlar genellikle bir sağlık sorununa yol açmazken, bir sonraki nesle aktarıldığında dengesiz bir hale gelebilir. Bu dengesiz translokasyonu alan sonraki nesillerde eksik veya fazla olacak şekilde kromozomal değişiklikler görülür.
Cri du chat kromozom 5’in kısa kolunda (p) kısmi bir eksikliğe (monozomi) bağlı gelişen bir sendromdur. Bütün kromozomlar kısa bir kola (p) ve uzun bir kola (q) sahiptir.
Araştırmacılar semptomların kromozom 5’teki özel bölgelerde ve kısa kolda olan eksikliklere bağlı olduğunu düşünmüşlerdir ve buna bağlı olarak bazı araştırmalar yapmışlardır. Bu araştırmalar ışığında bazı genlerin cri du chat oluşumunda etkili olduğunu düşünmektedirler.
Semaphorin F geni 5p15.2 lokasyonunda bulunur ve semptomlarda çeşitliliğe sebep olabilir.
D-catenin (CTDNN2) geni eksikliği de 5p15.2 lokasyonunda bulunur ve ciddi zihinsel gerilikle ilişkilendirilmiştir. Bu protein erken sinirsel gelişim sırasında eksprese edilir.
Birçok vaka rastgele bir şekilde meydana gelmektedir. Bu eksikliğe bağlı sendromların birçoğu (%80 -90) babaya bağlıdır, yani sperm kaynaklıdır.
Teşhis
Yenidoğanda kedi benzeri ağlamayla karakterize bir rahatsızlık olması sebebiyle, bu ağlamanın duyulmasının ardından karyotip yöntemleri kullanılarak kromozom 5’in kısa kolunda eksiklik olup olmadığı araştırılır. FISH (Floresan in situ hibridizasyon) yöntemiyle hastalığın tanısı doğrulanır. Karyotip çalışmalarında genellikle ebeveynlerden birisinde dengeli translokasyon saptanır. PCR yöntemiyle kedi benzeri ağlamaya sebep olan gen lokasyonları saptanabilir.
Ek tanı yöntemi olarak X-RAY cihazları ile iskelet sistemindeki anormallikler (skolyoz gibi) teşhis edilir.
Teşhis yöntemleri halen geliştirilmekte olup şu an için kısıtlıdır.
Tedavi
Bu rahatsızlığa sahip bireylerde tedavideki ilk yaklaşım, spesifik semptomların tedavisine yöneliktir. Genellikle multidisipliner bir ekiple (Ortopedistler, cerrahlar, kardiyologlar, nörologlar, diş hekimleri ve diğer sağlık çalışanlarından oluşan) bütün spesifik semptomlar üzerinde tedavi yöntemleri belirlenir.
Tedavide erken teşhis hastaların yaşam kalitelerinin yükselmesi ve toplum hayatına uyum sağlaması açısından çok önemlidir.
Beyin hasarı bulunan hastalar için ne yazık ki spesifik bir tedavi yöntemi bulunmamaktadır lakin rehabilitasyon programları ile hastalığa sahip bireylerin topluma adaptasyonu sağlanmaktadır.
Beslenmede zorluk yaşayan çocuklar (emmede ve yutmada zorluk gibi) doğumun ilk haftasında tedavi programlarına başlatılır.
Erken rehabilitasyon programları (konuşma terapisi, fiziksel terapi gibi) gecikmekte olan motor ve beyin gelişimi için önemlidir. Bazı çocuklarda sensörinöral duyma bozuklukları bulunabilir. Bu çocuklarda işitme muayenesi ve tedavileri uygulanır.
Aşılama takvimlerinde bir sıkıntı bulunmaz. Tüm olması gereken aşılar önerilmektedir.,
Emery-Dreifuss Musküler Distrofisi (EDMD), kalp hastalığı, kontraktür ve kas atrofisi (zayıflığı) ile ilişkilendirilen nadir genetik bir hastalıktır. Adını 1960’larda bir Virginia ailesindeki bozukluğu ilk kez tanımlayan doktorlar olan Alan Emery ve Fritz Dreifuss’tan almıştır.
Kalıtım Paterni/Deseni
EDMD, çoğu durumda X’e bağlı çekinik ve otozomal dominant bir özellik olarak kalıtılabilir. Otozomal çekinik kalıtım son derece nadirdir, ancak en az bir ailede bildirilmiştir. EDMD’ nin otozomal dominant ve resesif formlarında erkekler ve dişiler eşit olarak etkilenirken, X’e bağlı formda öncelikle erkekler etkilenir ve kadın taşıyıcılar arasında bazı hastalık belirtileri görülür.
Belirti ve Semptomlar
EDMD’nin başlangıç yaşı, şiddeti ve ilerlemesi, aynı ailenin bireyleri arasında bile vakadan vakaya büyük ölçüde değişiklik gösterir. Etkilenen bazı bireyler, çocukluk çağında hızlı hastalık ilerlemesi ve ciddi komplikasyonlar yaşayabilir; diğerleri yetişkin başlangıçlı ve yavaş ilerleyen bir seyir gösterebilirler.
EDMD klinik olarak üç semptom ile karakterize edilir: çocuklukta başlayan ve ilk olarak dirsekleri, ayak bileklerini ve boynu etkileyen eklem kontraktürleri; kas atrofisi; ve kalp yetmezliği, çarpıntı, iletim bozuklukları gibi kardiyak belirtiler. Semptomlar normal olarak yaşamın ikinci on yılında veya sonrasında ortaya çıkar. Kas atrofisi geç çocukluk ya da erken ergenlikte genellikle üst kollarda ve alt bacaklarda gelişir. Kas atrofisinin ilerlemesi, sonradan hızlanma eğilimindedir ancak yaşamın ilk otuz yılında genellikle yavaştır. Otozomal dominant EDMD’li bazı kişiler sonunda yürüme yeteneğini kaybedebilir. X’e bağlı EDMD’de yürüme kaybı nadirdir. Kontaktürler, kas atrofisinden önce meydana gelirler ve ergenlik döneminde, daha belirgin durumdadırlar. Bazı EDMD’li hastalarda solunum kaslarının zayıflığından dolayı solunum yetmezliği görülür. EDMD’nin üçüncü belirgin özelliği olan kalp anormallikleridir. Başlangıcı değişebilse de, genellikle yaşamın ikinci on yılından sonra gelişir. Kadın taşıyıcılar hiçbir zaman kas semptomları veya belirtileri göstermezler, ancak %20’sinde kalp hastalığı gelişebilir.
Genetik Değişiklikler/Etken Faktörler
EDMD, her bir kas hücresinin çekirdeğini çevreleyen nükleer zarda protein üreten genlerdeki mutasyonlardan kaynaklanır.
EDMD’nin X’e bağlı formu, X kromozomunun (Xq28) uzun kolunda bulunan EMD (STA olarak da bilinir) geninin bozulması veya mutasyona uğramasından kaynaklanır. EMD geni, emerin adı verilen bir kas proteinini kodlar. Emerin vücudun çoğu hücre tipinde bulunur ancak özellikle iskelet ve kalp kasları yüksek ekspresyon seviyelerine sahiptir.
EDMD’nin otozomal dominant ve otozomal resesif formlarının, 1. kromozomun uzun kolunda (1q21.2) bulunan aynı genin mutasyonlarından kaynaklanır. Gen, LMNA geni olarak bilinir ve lamin A ve lamin C proteinlerini kodlar.
EDMD, aynı zamanda doğrudan emerin ile etkileşime giren nükleer zarf proteinleri nesprin-1 ve -2’deki mutasyonlardan da kaynaklanabilir. İskelet kasında yüksek seviyelerde eksprese edilen nesprinler, kas gelişimi sırasında çok çekirdekli kas hücrelerinde çekirdeklerin uygun şekilde konumlandırılmasını ve bağlanmasını sağlar.
Çekirdeği hücre iskeletine bağlamak için nesprinlerle bir kompleks oluşturan SUN alanı proteinleri SUN1 ve SUN2’deki mutasyonlar da EDMD’ye neden olabilir. Bazı EDMD vakalarında, emerine bağlanan bir nükleer membran proteini olan LUMA olarak da bilinen FHL1 genindeki mutasyonlardan kaynaklandığı belirlenmiştir.
Teşhis Yöntemleri ve Tedaviler
Emery-Dreifuss Musküler Distrofisi hastalarına teşhis konulurken önce doktor tarafından bir hasta ve aile öyküsü alınır ve fiziki muayene yapılır. Doktor ayrıca hastanın zayıflığının kaslardaki bir sorundan mı yoksa onları kontrol eden sinirlerdeki bir sorundan mı kaynaklandığını belirleyebilir. Genellikle, zayıflığın kaynağı fiziksel bir muayene ile belirlenebilir. Bazen sinir iletim çalışmaları ve elektromiyografi (EMG) adı verilen özel testler yapılır.
Ayrıca etkilenen deriden biyopsi örneği alınması, kas hücrelerinde hangi kas proteinlerinin bulunduğu ve bunların normal miktarlarda ve doğru yerlerde bulunup bulunmadığı hakkında bilgi sağlayabilir. EDMD’li hastalardan alınan deri biopsisinde derideki çekirdeklerde emerin proteininin yokluğunun gösterilmesi tanıyı kesinleştirir.
Ek olarak hastaya genetik testlerin yapılması ile EDMD’ye neden olan belirli gen mutasyonları analiz edilerek belirlenir, bu hastalığın olası seyrini tahmin etmeye yardımcı olabilir ve ailelerin hastalığı bir sonraki nesle geçirme riskini değerlendirmesine yardımcı olabilir.
Emery-Dreifuss Musküler Distrofisi’ne sahip hastaların semptomların iyileştirilemesi hastanın yaşam kalitesini arttırır. EDMD’li hastalarda meydana gelen kontraktürleri önlemek zordur, ancak fizik tedavi ile hareket açıklığını korumak gelişimlerini yavaşlatmaya yardımcı olabilir.
Ek olarak EDMD’li hastaların birçoğunda gözlemlenen kardiyomiyopati, vücuda kan pompalama yeteneğindeki bir bozulmadır. kardiyomiyopatinin düzeltilmesinde bazı ilaçlar yardımcı olabilir.
Kaynaklar
Heller, S. A., Shih, R., Kalra, R., & Kang, P. B. (2020). Emery-Dreifuss muscular dystrophy. Muscle & nerve, 61(4), 436–448.
Madej-Pilarczyk A. (2018). Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus (Austin, Tex.), 9(1), 268–274.
Del Valle Sanchez, M. (2016). Emery-Dreifuss muscular dystrophy type 2: New de novo mutation in the lamin A/C gene. Neurologia (Barcelona, Spain), 33(8), 554-555.
‘Bunların neredeyse yarısı beş yaşına gelmeden yaşamını da yitiriyor.’
Hazal Sena ÇELEBİ
Öncelikle şunları sormak istiyorum: Nadir hastalıklar nedir? Nadir hastalıklar ile ilgili önemli sorunlar nelerdir?
Öncelikle duyarlılığın için çok teşekkür etmek istiyorum. Nadir hastalıklar, adından da anlaşılacağı gibi toplumda nadir gördüğümüz, sıklık olarak bakarsak da 2000 kişiden daha azında görülen -böyle bir tanım var- hastalıklardır. Tabii yöreden yöreye, bölgeden bölgeye bu hastalıklar değişebiliyor. Çünkü, her yörenin ayrı bir rahatsızlığı var. Bunlardaki ana sorunu, ben bir “ Çocuk Romatolog”u olarak yanıtlamak istiyorum. Bu hastaların hastaneye, hekime, tedaviye ulaşabilme zorlukları, tanınabilirlik zorlukları ve diğer bulgularla birlikte geliyor. En önemli özelliği de bunlardır. Sizin de çalışmış olduğunuz genetik kökenli hastalıklar ve bu genetik kökenli hastalıkları gerçekten tanımak çok zor. Bunlarla birlikte uğraşmak, bunları takip etmekte çok zor. Farklı klinik bulgularda daha az gördüğümüz, toplumda görmeye alışık olmadığımız ortamlarda hep aklımıza gelmesi gereken bir noktadır burası.
2. Nadir hastalıklar genellikle çocukları mı etkiliyor?
Tabii, her yaşta görülebiliyor; böyle bir genelleme yok. Ama çoğunlukla genetik kökenli oldukları için bunların birçoğu küçük yaşlarda ortaya çıkıyor. Doğumla birlikte başlıyorlar. Doğumla birlikte başladıklarında da bu çocuklar doğdukları anda çeşitli bulgularla, çeşitli verilerle karşımıza gelebiliyorlar ve bunlar karşımıza geldiği zaman gerçekten ortaya çıkan belli noktalar oluyor çocukluk çağında. Bunların neredeyse yarısı beş yaşına gelmeden yaşamını da yitirebiliyor. Bu nadir hastalıkların çoğu, yaşamla da bağdaşamıyor ve ne yazık ki çocuklarımızı kaybediyoruz. Aslında bunları daha erken tanıyabilmiş olsaydık daha çabuk tedavilerini sağlayabilirdik. Ama bu, tek bir hekimin yapabileceği ya da tek bir kişinin yapabileceği bir şey gibi değil. Yani birçok insanın iç içe yapıp birçok insanın iç içe değerlendirebileceği bir durum olarak karşımıza geliyor.
3. Özellikle size sormak istediğim sorulardan birisi: Çocukluk çağı romatizmal hastalıkları ve belirtileri nelerdir? Nadir olanlar özellikle.
Çok teşekkür ederim bu soru için. Çocukluk çağı romatizmal hastalıkları deyince gerçekten çok geniş bir kavramla karşı karşıyayız. Ve bu geniş kavramın içinde birçok veri karşımıza çıkıyor. Ne zaman düşünmeliyiz bunları? Bir kere ağrı hissetmek, hepimizin yaşadığı bir şey. Ama o ağrı dışında; hareket kısıtlılığı, yürüme zorluğu, eklemlerde şişlik ve kızarıklık gibi bulgular. Ya da bir çocuğun yaptığı en önemli eylem biliyorsunuz ki yaramazlıktır. Çocuğun yaramazlık yapamaması, yaramazlığını kaybetmesi, hareketlerini yapamaması çok önemli bir veri olarak karşımıza geliyor ve bunlarla birlikte ortaya çıkan bir durum oluyor. Onun dışında kontrol altına alınamayan ateşler, özellikle değişik cilt döküntüleri gibi bulgular aklımıza çocukluk çağı romatizmal hastalıklarını getirmeli. Tabii bunlar içinde ana grup olarak otoinflamatuar dediğimiz bir geniş grup vardır. Bunlar genetik geçişli olan hastalıklar ve genetik geçişli olduğu için de bunlar çocukluk çağında daha sık bulgularla birlikte karşımıza çıkabiliyor Hazal.
Kesinlikle yaşanıyor. Bize ulaşmada, ilgili merkezlere ulaşmada bazen sıkıntılar oluyor. Ama bu sıkıntıları ortadan kaldırdığımız anda bunların birçoğunu da ortadan kaldırmış oluyoruz kolaylıkla. Yani hastalar bize ulaştığı anda daha sağlıklı olarak tedavilerini düzenlemiş oluyoruz ve bunlarla birlikte gidiyoruz. Tabii ki geldikten sonrası kolay. Ama ilaca erişimdeki sıkıntılar gibi birçok şeyi de bizler çabalayarak çözmeye çalışıyoruz.
5. Hasta ve hasta yakınlarının yaşamında erken tanı, doğru tedavi ve düzenli takibin önemi nedir?
Üzerine de vurguladığın gibi Hazal, gerçekten çok önemli. Her hastayı ne kadar erken tanıyabilirsek o kadar başarılı oluyoruz tedavide. Yani erken tanımamız, erken olarak bunları değerlendirmemiz çok önemli. Tedaviye erken başladığımız zaman, erken tanıdığımız zaman -hastalıkta oluşabilecek iki ana grubumuz var. Birinci grup, hastaları aktif tutmak istemiyoruz; ikinci grup, hasar oluşmasın istiyoruz.- yani bizim en büyük korkumuz çocukların bu hastalıklardan ötürü hasar almaları. Yani hasar aldıkları zaman ciddi olarak etkilenmeler oluyor. Eklemleri etkileniyor, ortalama ömürleri 80 yıl olduğu için. Yani biz var olan eklemlerimizi 80 sene kullanabileceğiz demek bu. Bunlarda oluşan bir hasar, geriye dönüşsüz olabiliyor. Ve bu hasarın önlenmesi için çabalıyoruz, bütün amaçlarımız da bunun için.
6. Nadir hastalıkların -özellikle sizin branşınız için soruyorum- tedavisi mümkün mü?
Tabii ki tanıya göre değişiyor. Bu ülkede sağlıkçılar, sağlıkla uğraşanlar; işte bunlar genetikçiler, biyologlar, doktorlar, hemşireler… bütün sağlık çalışanlarının tek bir amacı var: Yaşamları boyunca, eğitimleri boyunca toparlamış oldukları bu iyiliği taşımak. İyiliği taşımak için çabalıyorlar, iyiliği taşımak için uğraşıyorlar. Ve iyiliği taşımak için yaptıkları çabalarla birlikte yürütüyorlar bu olayları. Ve bunlarla birlikte karşımıza çıkan bir bulgu oluyor. Ve bu yüzden biz hastaların birçoğunu tedavi etmeye çalışıyoruz. Ama bu spesifik olarak hastalıktan hastalığa değişiyor. Her hastalığın farklı bulguları, farklı verileri olabiliyor. O yüzden de hastalık bazında konuşmak daha doğru oluyor. Hepsine elimizden geleni yapmaya çalışıyoruz. Yarısını da tam olarak tedavi edebiliyoruz diyelim.
7. Nadir hastalıklarla savaşan hastaların ve hasta yakınlarının günlük hayatta yaşadığı zorluklar nelerdir, biraz bahsedebilir misiniz?
Gerçekten çok zor bir hayat. Düşünün hep hasta bir çocuğunuz olduğunu ya da hep hasta olduğunuzu… gerçekten yaşanması kolay bir şey değil. Tabii ki sıkıntılar çok büyük ve bunlara elimizden geldiğince bizler de yardımcı olmaya çalışıyoruz, bunların hepsini düzeltmeye çalışıyoruz. Bunların hepsini düzenlemeye ve toparlamaya çalışıyoruz. Gerçekten zorlu bir iş, bu hastalarımıza mutlaka hasta dernekleri aracılığıyla, diğer aracılarla da yardımcı olmak zorundayız. Ve bunlarla birlikte bunları toparlamak bizler için de rahatlatıcı bir etki yaratacaktır.
8. Az önce de derneklerden bahsettiniz. Nadir hastalıklar için kurulan dernekleri ve düzenlenen farkındalık projelerini yeterli buluyor musunuz -ülkemiz için-?
Bizim ülkede ben şu konudan adım gibi eminim: uzmanlık dernekleri, özellikle UDEK çatısı altında toplanan bütün uzmanlık dernekleri ellerinden gelen iyiliği hastalara taşımak için uğraşıyorlar. Ben” Çocuk Romatoloji Derneği” başkanıyım. Çocuk Romatoloji Derneği’nde ben ve arkadaşlarım, bütün çabamız hastalarımızın iyiliği için. Elimizden geleni gösteriyoruz. Yeterli mi? Tabii ki değil. Daha çok şey yapmamız gerekiyor. Hastalarımızı daha çok bilgilendirmemiz, ailelerimizi daha çok bilgilendirmemiz gerekiyor. Ve bunlarla birlikte güzel bir geleceğe çocuklarımızı taşımamız gerekiyor. Ama yaptığımız hiçbir şey hala yeterli değil. Hala daha çok yapacak işimiz var.
9. Son olarak eklemek istediğiniz bir husus var mı?
Hiçbir şey yok. Çalışmaya devam edin. Bence çalıştıkça her şey çok güzel olacak. Gerçekten bu ülkenin gençlerinden ben çok ümitliyim. Çok güzel şeyler yapacağınıza da adım gibi inanıyorum.
Kistik fibroz-gastrit-megaloblastik sendromu ilk olarak akraba olan iki arap ebevyinin iki çocuğunda ortaya çıkmıştır. Bu hastalık genel olarak anemi( kanda azalmış hemoglobin), megaloblastik anemi( kötücül anemi), gastrit(mide yanması), helikobakteri pilori, folik asit eksikliği(Vitamin B9 eksikliği) ve zihinsel yetersizlikle karakterizedir. Ama bu nadir hastalık hakkında 1991’den beri daha fazla araştırma yapılmamış ve literatüre yeni bilgiler eklenmemiştir. Bu yüzden bu hastalık hakkındaki bilgiler bu sayfadakilerle sınırlı olup yeni araştırmaların literatüre kazandırılması beklenmektedir.
Semptomlar
Uzun yüz, alın kısmında büyüme, kepçe kulak, küçük ve derinde olan gözler, alt çenenin yetersiz gelişmesi, tekrarlayan enfeksiyonlar, gastrit, ishal, çok terleme, zeka geriliği, metabolizmik anemi, folik asit eksikliği gibi çok çeşitli ve vücudun her bir alanınan etki edebilen semptomları bulunmaktadır. Ayrıca siroz, akciğer hastalıklarına bağlı gelişen kalp hastalıkları, safra yolu tıkanıklığı gibi bir çok hastalığın kapısını aralamaktadır.
Görülme Sıklığı ve Kalıtım Geni
Bunun hakkında şimdiye kadar yalnızca bir araştırma yapılmış. Bu da gösteriyor ki bu hastalık oldukça nadir bir hastalıktır.
Ek olarak, bu hastalık otozomal çekinik olarak kalıtılır. Yani eşey hücreleriyle değil de vücut hücreleri tarafından kalıtılmaktadır. Çekinik olması görülme sıklığının azalmasında önemli bir etkendir.
Uzuv-kuşak kas distrofisi hastalığı kol ve bacaktaki kasların zayıflaması sonucu ortaya çıkan bir hastalık olarak tanımlanabilir. Proksimal kaslar diğer bir deyişle vücuda en yakın olan kaslar örneğin omuz, üst kol, pelvik ve uyluk kasları bu hastalıktan en çok etkilenen kas gruplarıdır. Uzuv kuşağı kas distrofisi hastalığının şiddeti, hastalığın ortaya çıkış yaşı, ve hastalığın diğer karakteristik özellikleri kişiden kişiye göre birçok farklılıklar gösterir. Aynı aileden olup bu hastalığa sahip olan kişilerde bile bu özellikler çok farklı olabilir. Uzuv kuşağı kas distrofisi hastalığının belirtileri belirli bir yaştan sonra kendini göstermeye başlar.Bazı kişilerde bu belirtiler hafif durumda kalabilirken çoğu kişide bu belirtilerin giderek şiddetlendiği gözlemlenir. Uzuv kuşağı kas distrofisinin ilk evrelerinde etkilenen hastalar yürümekte ve koşmakta zorluklar çekebilir. Bu hastalık nedeniyle çömelip kalkma gibi hareketleri gerçekleştirirken dışardan destek almaları gerekebilir. Hastalığın ilerleyen aşamalarında bireyler ayakta durma, yürüme, koşma, kollarını kullanabilme gibi yetkinliklerini tamamen kaybederek tekerlekli sandalye kullanmaya başlarlar. Bu hastalıkta kas kaybı sonucu bireylerin duruşunda ve omuzlarının görünüşünde farklılık ortaya çıkar. Özellikle zayıf omuz kaslarının sonucu olarak kürek kemikleri arkaya doğru çıkar bu durum skapular kanatlanma olarak adlandırılır. Ayrıca bu hastalıktan etkilenen kişilerde kavisli bir bel veya yana doğru kıvrılan diğer bir adıyla skolyoz olarak adlandırılan omurga ortaya çıkabilir. Bazı kişilerin kalçalarında, yüzlerinde ayak bileklerinde, ve dirseklerinde hareketi kısıtlayan eklem sertliği olarak adlandırılan sorun ortaya çıkabilir. Baldırdaki kasların çok fazla büyümesi de bu hastalığa sahip bazı bireylerde gözlenen bir durumdur. Kardiyomiyopati olarak adlandırılan kas kaslarının zayıflaması bu hastalığın bazı türlerinde görülebilen bir durumdur. Bu hastalıktan etkilenen bazı bireylerde nefes almak için kullanılan kasların zayıflaması sonucu soluk alıp vermede problemler ortaya çıkar. Bu solunum sorunları bazı hastalarda çok şiddetli gözlemlenir bu durumda bu hastaların solunum cihazlarından destek alması gerekir. Uzuv kuşağı kas distrofisi genel olarak zekayı etkileyecek sorunlara neden olmaz fakat nadirde olsa bazı durumlarda gelişimsel gecikme ve zihinsel yetersizlik olduğu gözlemlenmiştir.
Semptomlar
Ayak parmakları üzerinde duramamak ve ayak parmakları üzerinde yürüyememek
Yürümekte zorlanmak
Destek almadan çömelip kalkma işini gerçekleştirememek
Koşmakta zorluk çekmek
Lordoz ve skolyoz gibi omurga eğrilikleri
Eklem sertlikleri
Teşhis
Mutasyon analizi ile yapılan çalışmalarda uzuv-kuşak kas distrofisi hastalığına sahip olan kişilerde kalpain-3 protein eksikliği olduğu gözlemlenmiştir. Bireylerde kalpain-3 eksikliği söz konusu olduğunda bu hastalığa sahip olma olasılığı oldukça yüksektir. Kalpain-3 proteini eksikliğinin görüldüğü hastalarda yapılan test sonuçlarına göre bu hastaların CAPN3 genlerinde mutasyonlara sahip olduğu gözlemlenmiştir.
Tedavi
Uzuv-kuşak kas distrofisi hastalığı için net ilaç tedavileri henüz yoktur. Gen tedavisi, hücre tedavisi, bazı farmakolojik tedavi denemeleri dahil olmak üzere farklı bir çok yaklaşım hayvan modellerinde ve deneysel çalışmalarda halen araştırılmaktadır. Uzuv-kuşak kas distrofisi hastalığı farklı türleri de dahil olmak üzere yetişkin kas distrofisi sorunlarında miyostasin için nötralize edici MYO-029 antikoru son zamanlarda gerçekleştirilen bir klinik çalışmada güvenilirlik ve tolere edilebilirlik özellikleri gösterdi (Guglieri et all., 2008). ABD Ulusal Sağlık Enstitüsü’ne göre bu hastalık için şu anda devam eden iki tan klinik çalışma vardır. Bu klinik çalışmalardan ilkinde yapılan hayvan deneyleri sonucunda fonksiyonel α-sarkoglikan proteini üretilerek kas gücünün arttığı gözlemlenmiştir (Rodino et all.,2008).
Kalıtım Paterni/Deseni
Uzuv kuşağı kas distrofisi farklı kalıtım türlerine sahip olabilir. Bu hastalığın çoğu türü otozomal resesif bir modelde kalıtılır. Bunun anlamı her hücredeki genin her iki kopyasınında mutasyona sahip olmasıdır. Eğer bir birey otozomal çekinik hastalığa sahipse bu o kişinin ebeveynlerinin mutasyona uğramış genlerinin birer kopyasını taşıdığı anlamına gelir.
Görülme Sıklığı
Uzuv-kuşak kas distrofisi hatalığının belirlenmesi oldukça zordur çünkü hastalığın özellikleri kişiden kişiye göre birçok farklılık gösterir ve bu hastalığın belirtileri başka kas hastalıklarıyla aynıdır. Tahminlere göre bu hastalığa sahip olma sıklığı 14.500’de 1 ile 123.000 kişide 1 arasında değişmektedir.
Spondilo “omurga”, metafiz “kemiğin büyüme plakasını (kemiğin çocukluk döneminde büyüyen kısmı)” içeren geniş kısmını ifade etmektedir. Displazi “anormal büyüme” anlamına gelmektedir. [1] Yaklaşık olarak kişi iki yaşındayken tespit edilen, yürüme ve büyüme bozuklukları ile ilişkili heterojen bozukluk grubu olarak değerlendirilen nadir genetik hastalıktır.[2]
Genetik Değişiklikler/Etken Faktörler
Spondilometafizyal Displazi birçok farklı tipte görülmekte olan bir nadir hastalıktır. Bu tipler belirti ve semptomlar bakımından ortak noktaları olsada farklılık gösterebilmektedir. Bazıları şu şekildedir:
Spondilometafizyal Displazi, Kozlowski tipi gövdeyi tutan boy kısalığı ile karakterize olan bir otozomal dominant kemik hastalığıdır. Genellikle erken çocuklukta, düzensiz boy ile zayıf büyüme ve eğik bacaklı olma durumundan kaynaklı olarak paytak yürüyüş fark edildiğinde başlamaktadır. Eklemlerin erken osteoartriti (kireçlenme) de yaygındır. Küçük eller ve parmaklar, omurga deformiteleri ve kısa omu, hafif metafiz değişiklikleri, kemikleşmede ciddi gecikme, kare, kısa, alevlenmiş iliak kanatlar (pelvik kemiğinin en geniş kısmı) görülmektedir.[3]
Spondilometafizyal Displazi Cezayir Tipi
Diğer isimler:
Japon tipi spondilometafizyal displazi;
Schmid metafizyal disostoz;
Spondilometafizyal displazi Schmidt tipi;
Şiddetli genu valgum ile birlikte spondilometafizyal displazi
Boy kısalığı, miyop, küçük pelvis, ilerleyici kifoskolyoz, bilek deformitesi, şiddetli genu valgum, kısa uzun kemikler ve orta dereceli spinal değişiklikler ve el ve ayaklarda minimal değişikliklerle birlikte şiddetli metafizyal displazi ile karakterizedir.[4]
Spondilometafizyal Displazi Köşe Kırığı Tipi
Diğer isimler:
Spondilometafizyal displazi Sutcliffe tipi;
Sutcliffe SMD’si;
Sutcliffe tipi spondilometafizyal displazi
Boy kısalığı, gelişimsel koksa vara, progresif kalça deformitesi, uzun tübüler kemiklerin simüle “köşe kırıkları” ve vertebra gövdesi anormallikleri ( çoğunlukla oval vertebra gövdeleri) ile ilişkili bir iskelet displazisidir. Metafiz köşe kırığı, uzuv ağrısı, ortak hareketliliğin sınırlandırılması, yüzün sağ ve sol tarafları arasında asimetri, yüksek damak, sivri çene, şaşılık, diş anormalliği, kırık oluşumuna karşı duyarlılığın artması, çıkıntılı kulak, demir eksikliği anemisi gözlenmektedir. [5]
Spondilometafizyal Displazi A4 Tipi
Orantısız boy kısalığı, şiddetli femur boyun deformitesi, belirgin metafiz anomalileri ve ön dil benzeri deformiteye sahip ovoid vertebra gövdelerinden oluşan platispondili ile karakterize edilen nadir görülen bir primer kemik displazisi bozukluğudur.[6]
Spondilometafizyal Displazi Sedaghat Tipi
Diğer isimler:
Ölümcül metafiz displazisi.[7]
Hafif ekstremite kısalması, platispondili, gecikmiş epifiz ossifikasyonu, düzensiz iliak krestleri ve pulmoner kanama ile karakterize ciddi metafiz kondrodisplazisi ile karakterize nadir görülen ölümcül bir hastalıktır. Etkilenen bebeklerde şiddetli hipotoni ve kardiyorespiratuar problemler görülür; çoğu solunum yetmezliği nedeniyle doğumdan sonraki günlerde ölür. Kardiyak anormallikler arasında iletim kusurları, tam kalp bloğu ve yapısal anomaliler bulunur. SMDS’li bebeklerin yarısında, korpus kallozum agenezisi, belirgin frontotemporal pakigiri, basitleştirilmiş giral patern, kısmi lizensefali ve şiddetli serebellar hipoplazi dahil olmak üzere anormal nöronal göç ile uyumlu merkezi sinir sistemi malformasyonları olduğu bildirilmektedir.[8]
Diğer isimler:
Spondilometafizyal displazi Richmond tipi
Şiddetli boy kısalığı, torakolomber kifoskolyoz ve kontraktürlü genişlemiş eklemlerle karakterize, nadir görülen bir primer kemik displazisi hastalığıdır. Psikomotor gecikme ve zihinsel engellilik de ilişkili olabilmektedir. Radyografik özellikler arasında düz vertebra gövdeleri, uzun kemiklerin ve iliak kretlerin metafizlerinin dantelli kemikleşmesi ve kafa tabanının belirgin sklerozu yer almaktadır.[9]
Spondilometafizyal displazi koni- çubuk distrafisi, iletimi otozomal çekinik olan bir nadir genetik hastalık tipidir.[10] Bu tipte retina incelmesi, görme bozukluğu, kornea berraklığında azalma, nistagmus (istemsiz, hızlı, ritmik göz hareketleri), zihinsel engellilik gibi belirtileri görülmektedir.[11]
Belirti ve Semptomlar
Bozukluklar, şiddetli kısa boy, omurga kolonunun anormalliği, orantısız kısa boy, koksa vara (boyun ile femur şaftı arasındaki açının 120° altına düşmesi), yürüme bozukluğu, genu varum (parantez bacak), brakidaktili (parmak kısalığı), genu valgum (X bacak), kifoz (omurganın öne eğrilmesi), platispondili (düzleşmiş omurlar), miyop, skolyoz (omurganın yana eğriliği), belirgin kalça ve diz metafiz lezyonları ile karakterize edilmektedir. Farklı spondilometafiz displazisi formları bulunmaktadır. Bunlar etkilenen metafizlerin tutulumunun lokalizasyonu ve ciddiyeti ile ayırt edilebilmektedir. En yaygın form Kozlowski tipi spondilometafizyal displazidir. ‘Köşe kırığı’ veya Sutcliffe tipi olarak adlandırılan spondilometafizyal displazi formu, çok şiddetli koksa vara ile sonuçlanır. Daha nadir bir formda (Cezayir veya Schmidt tipi) diz tutulumu daha baskın görünmektedir. Spondilometafizyal displazi, fasiyal dismorfizm ve dentinogenez imperfekta gibi diğer klinik belirtilerle birlikte de ortaya çıkabilmektedir.[2]
Genetik Görülme Sıklığı
Prevalansının 1/100.000 civarında olduğu tahmin edilmektedir.[2]
Kalıtım Paterni/Deseni
Hastalığın birçok tipi olması sebebiyle kalıtım paterni farklılık göstermektedir. Kozlowski tipi spondilometafizyal displazi, otozomal dominant bir şekilde ve ayrıca ‘köşe kırığı’ veya Sutcliffe tipi ve Cezayir (veya Schmidt) tipi olarak adlandırılan spondilometafizyal displazi şeklinde iletilir. Bazı ılımlı formların da genellikle otozomal dominant özellikler olarak aktarıldığı görülmektedir. Tip A4 ve retinitis pigmentosa ve optik atrofi ile ilişkili bir eksenel tip dahil olmak üzere çeşitli otozomal resesif formlar da tanımlanmıştır. X’e bağlı iletimi olan bir spondilometafizyal displazi formu da bildirilmiştir.[2]
Teşhis Yöntemleri ve Tedaviler
Teşhis:
Boyun, omurga, alt ekstremiteler ve pelvisin röntgeni
Kordun sıkıştırılıp sıkıştırılmadığını değerlendirmek için omuriliğin MRI taramaları
Kıkırdak değerlendirmek için kalçalara bir boya enjekte edilen artrogramlar
Tedavi:
Spondiloepifizyal displazi tedavisi, ilişkili ortopedik koşullara bağlı olarak değişir ve şunları içerebilir:
Servikal füzyon ve servikal omurganın (boyun) olası dekompresyonu – halo ve yelek gerektirebilir
Erken yakalanırsa skolyoz ve kifoz için destek
Skolyoz ve kifoz için spinal füzyon
Yanlış hizalamayı ve/veya subluksasyonu düzeltmek için kalça osteotomisi (kısmi çıkık ve kalça fleksiyon kontraktürleri)
Ayak deformiteleri için döküm
Özelleştirilmiş toplam eklem değiştirmeleri.[12]
Hastalıkla İlişkili Genler
Hastalık Tipi
İlişkili Gen
Spondilometafizyal displazi, Kozlowski tipi
TRPV4
Spondilometafizyal displazi Cezayir tipi
COL2A1
Spondilometafizyal displazi köşe kırığı tipi
FN1
Spondilometafizyal displazi Sedaghat tipi
GPX4
Spondilometafizel Displazi, Pagnamenta Tipi
PRKG2
Spondiloepimetafizal Displazi, Strudwıck Tipi
COL2A1
Knıest Displazi
COL2A1
Spondilometafizel Displazi, Megarban-Dagher-Melkı Tipi

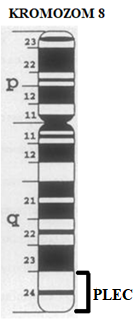 [9]
[9]

{kind=link}
{kind=link}
{kind=link}
{kind=link}