Ege Efecan ATASOY
Genel Bilgi
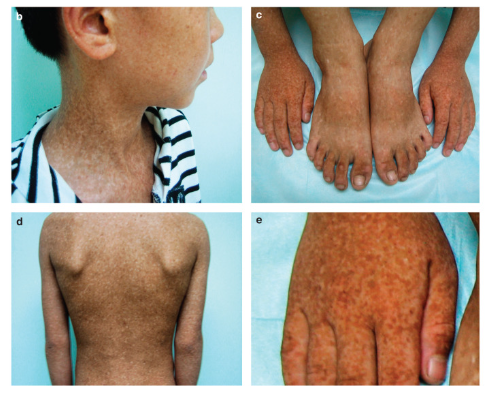
Cri-du-chat sendromu doğuştan itibaren hastanın büyüme ve gelişmesini olumsuz yönde etkileyen bir rahatsızlıktır. Bu rahatsızlığa sahip olan yenidoğanlarda sıklıkla tiz bir kedi benzeri ağlama, mikrosefali ve karakteristik yüz özellikleri (hipertelorizm, düşük kulaklar, küçük bir çene ve yuvarlak bir yüz gibi) görülür. Solunumda zorlanma ve beslenmede zorluk görülebilir. Bu hastalığa sahip bireylerde zihinsel bir engelin yanında gelişimde ve konuşmada gecikme görülür, ayrıca davranış bozuklukları da görülebilmektedir. Bu sendrom kromozom 5’in p kolu denilen bölgesindeki bir eksikliğe bağlı olarak görülmektedir. Genellikle semptomların şiddeti kromozom 5’te bulunan eksikliğin bölgesine ve büyüklüğüne bağlıdır. Bu eksiklik embriyonun erken aşamalarında meydana gelir ve genellikle ailesel bir kalıtıma bağlı değildir.

Semptomlar
1- Anormal ses
Çok sık görülmektedir.
2- Kedi-benzeri ağlama
Hastalığa ismini veren bu semptom oldukça tiz bir kedi benzeri ağlama ile karakterizedir.
Çok sık görülmektedir.
3- Epikantus
Epikantus ya da epikantal kıvrım, göz çevresinde bulunan kıvrılmış deri parçasıdır. Kıvrımın kirpikleri içe döndürüp göze zarar vermeye başladığı durumlarda tedavi edilir. Tedavi edilmemesi durumda başka hastalıklara sebebiyet verebilir.
Çok sık görülmektedir.
4- Ciddi seviyede zihinsel gerilik
IQ seviyesi 20 ila 34 arasında değişmektedir.
Çok sık görülmektedir.
5- Mikrosefali
Baş çevresinin yaş ve cinsiyete göre ortalamanın altındaki iki standart sapmadan daha küçük olduğu nörogelişimsel bir bozukluktur.
Çok sık görülmektedir.
6- Mikroretrognati
Alt çenenin geride olması durumu.
Çok sık görülmektedir.
7- Yuvarlak yüz
Çok sık görülmektedir.
8- Ciddi düzeyde gelişim eksikliği
Çok sık görülmektedir.
9- Aşağı eğimli palpebral fissürler
Gözlerin dış köşelerinin aşağıya doğru meyilli olduğu durum.
Sıklıkla görülmektedir.
10- Hipertelorizm
Gözler arasındaki mesafenin abartılı bir seviyede artması.
Sıklıkla görülmektedir.
11- İntrauterin gelişme geriliği
Anne rahmindeki bebeğin, gebelik haftasına göre olması gerekenden küçük olmasıdır.
Sıklıkla görülmektedir.
12- Skolyoz
Spinal deformiteler içerisinde en sık karşılaşılan ve ilerleyen evrelerde son derece ciddi duruş bozukluklarına neden olan üç boyutlu bir omurga deformitesidir. Bu deformite omurganın yapısal bozukluğundan kaynaklı olarak ortaya çıkabileceği gibi omurga dışı sebeplere bağlı olarak da gelişebilir.
Sıklıkla görülmektedir.
13- Kısa boyun
Sıklıkla görülmektedir.
14- Küçük el
Sıklıkla görülmektedir.
15- Kardiyovasküler sistem morfolojisinde anormallikler
Kalp ve damarlarda yapısal bozukluklar.
Zaman zaman görülmektedir.
16- Kasık fıtığı
Karın içindeki organların (ince bağırsaklar, bağırsak yağları gibi), karın duvarındaki zayıf bölgelerden çıkarak cilt altında şişlik oluşturmasıdır.
Zaman zaman görülmektedir.
17- Eklem hipermobilitesi
Eklemlerin normalden çok daha fazla hareket aralığının bulunması durumu. Bu rahatsızlığa sahip kişiler eklemlerinin fazla hareketine bağlı ağrı çekebilirler.
Zaman zaman görülmektedir.
Görülme Sıklığı
Prevalansının 15.000’ de 1 ila 50.000’de 1 olduğu düşünülmektedir. Toplumda tanısı konulmamış hastaların varlığı sebebiyle bu aralık konusunda kesinlik vermek zordur. Görülme sıklığındaki başka bir önemli detay ise kadın hastaların oranı erkek hastalardan daha yüksektir.
Genetik Paterni/Deseni
Çoğu vakada ailesel bir kalıtım görülmez. Eksiklik, ya da silinme, çoğu ya üreme hücrelerinin(sperm ya da yumurta) gelişimi sırasında ya da embriyonun erken gelişimi sırasında rastgele bir şekilde meydana gelir.
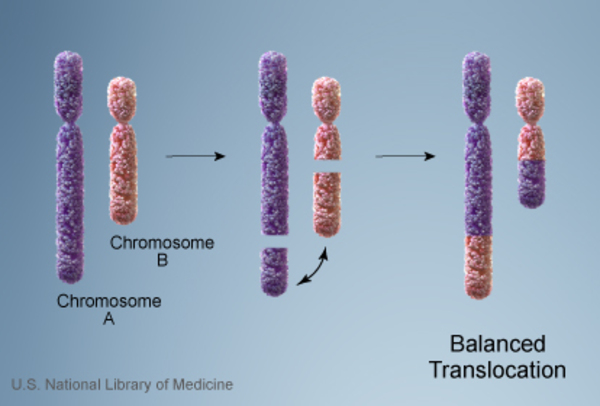
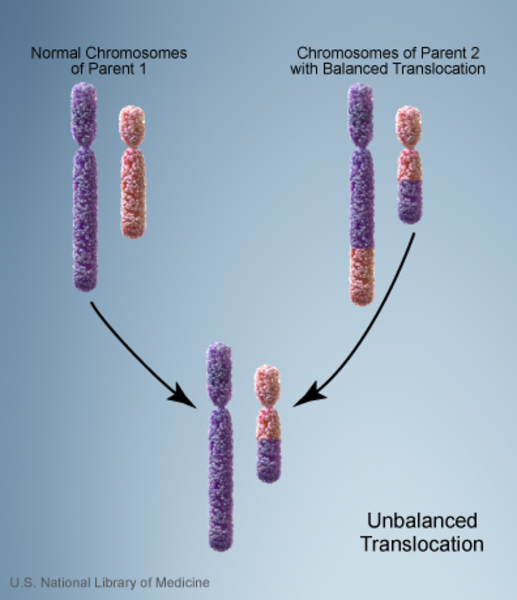
Vakaların %10’unda kromozom anormalliği hasta olmayan bir ebeveynden gelmektedir. Bu tarz durumlarda ebeveyn dengeli translokasyon adı verilen bir kromozomal yeniden düzenlemeye sahiptir. Dengeli translokasyonlar genellikle bir sağlık sorununa yol açmazken, bir sonraki nesle aktarıldığında dengesiz bir hale gelebilir. Bu dengesiz translokasyonu alan sonraki nesillerde eksik veya fazla olacak şekilde kromozomal değişiklikler görülür.
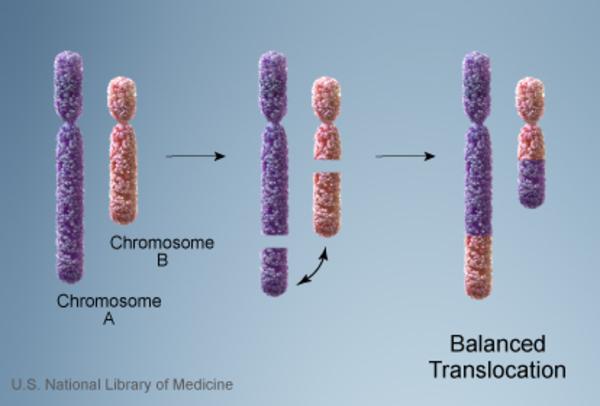
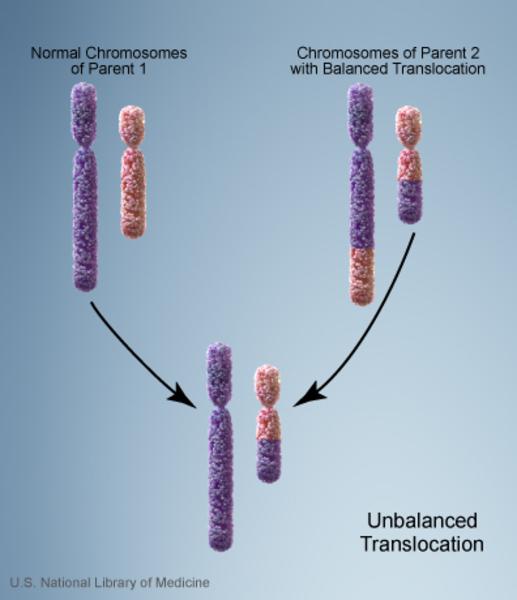
Cri du chat kromozom 5’in kısa kolunda (p) kısmi bir eksikliğe (monozomi) bağlı gelişen bir sendromdur. Bütün kromozomlar kısa bir kola (p) ve uzun bir kola (q) sahiptir.
Araştırmacılar semptomların kromozom 5’teki özel bölgelerde ve kısa kolda olan eksikliklere bağlı olduğunu düşünmüşlerdir ve buna bağlı olarak bazı araştırmalar yapmışlardır. Bu araştırmalar ışığında bazı genlerin cri du chat oluşumunda etkili olduğunu düşünmektedirler.
- Telomeraz reverse transkriptaz geni kromozom 5’in 13.33 kısa kolunda (5p13.33) lokalizedir.
- Semaphorin F geni 5p15.2 lokasyonunda bulunur ve semptomlarda çeşitliliğe sebep olabilir.
- D-catenin (CTDNN2) geni eksikliği de 5p15.2 lokasyonunda bulunur ve ciddi zihinsel gerilikle ilişkilendirilmiştir. Bu protein erken sinirsel gelişim sırasında eksprese edilir.
Birçok vaka rastgele bir şekilde meydana gelmektedir. Bu eksikliğe bağlı sendromların birçoğu (%80 -90) babaya bağlıdır, yani sperm kaynaklıdır.
Teşhis
Yenidoğanda kedi benzeri ağlamayla karakterize bir rahatsızlık olması sebebiyle, bu ağlamanın duyulmasının ardından karyotip yöntemleri kullanılarak kromozom 5’in kısa kolunda eksiklik olup olmadığı araştırılır. FISH (Floresan in situ hibridizasyon) yöntemiyle hastalığın tanısı doğrulanır. Karyotip çalışmalarında genellikle ebeveynlerden birisinde dengeli translokasyon saptanır. PCR yöntemiyle kedi benzeri ağlamaya sebep olan gen lokasyonları saptanabilir.
Ek tanı yöntemi olarak X-RAY cihazları ile iskelet sistemindeki anormallikler (skolyoz gibi) teşhis edilir.
Teşhis yöntemleri halen geliştirilmekte olup şu an için kısıtlıdır.
Tedavi
Bu rahatsızlığa sahip bireylerde tedavideki ilk yaklaşım, spesifik semptomların tedavisine yöneliktir. Genellikle multidisipliner bir ekiple (Ortopedistler, cerrahlar, kardiyologlar, nörologlar, diş hekimleri ve diğer sağlık çalışanlarından oluşan) bütün spesifik semptomlar üzerinde tedavi yöntemleri belirlenir.
Tedavide erken teşhis hastaların yaşam kalitelerinin yükselmesi ve toplum hayatına uyum sağlaması açısından çok önemlidir.
Beyin hasarı bulunan hastalar için ne yazık ki spesifik bir tedavi yöntemi bulunmamaktadır lakin rehabilitasyon programları ile hastalığa sahip bireylerin topluma adaptasyonu sağlanmaktadır.
Beslenmede zorluk yaşayan çocuklar (emmede ve yutmada zorluk gibi) doğumun ilk haftasında tedavi programlarına başlatılır.
Erken rehabilitasyon programları (konuşma terapisi, fiziksel terapi gibi) gecikmekte olan motor ve beyin gelişimi için önemlidir. Bazı çocuklarda sensörinöral duyma bozuklukları bulunabilir. Bu çocuklarda işitme muayenesi ve tedavileri uygulanır.
Aşılama takvimlerinde bir sıkıntı bulunmaz. Tüm olması gereken aşılar önerilmektedir.,
Hastalığın Diğer İsimleri
- 5p deletion syndrome
- 5p- syndrome
- Cat cry syndrome
- Chromosome 5p- syndrome
- Monosomy 5p
Kaynaklar



{kind=link}
{kind=link}
{kind=link}
{kind=link}