Rana ÖZDOĞAN
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Demodikozis, başta yüz bölgesi olmak üzere derinin belirli bölgelerini etkileyen nadir bir enfeksiyon hastalığıdır. Demodeks akarları olarak bilinen bir tür iç paraziti bu hastlığa sebep olmaktadır. Demodex folliculorum ve Demodex brevis olmak üzere vücutta iki türü bulunan bu parazitler herhangi bir genetik değişikliğe sebep olmamakla birlikte, hemen hemen her yaş grubundaki insanların cilt florasında bulunmaktadır. Özellikle, yanak, alın, burun kenarları ve kulak çevresi gibi yüz bölgelerinde yaygındırlar. Genelde zararsız durumdadırlar. Ancak, sayıları arttığı takdirde demodikozis adı verilen cilt rahatsızlığına sebep olmaktadır. Normalde yoğunluğu az olan Demodex parazitleri, sayıları arttığında bağışıklık sistemini bir parçası olan toll-benzeri reseptörleri (TLR) üzerinden etkileşime geçerek deride enfeksiyon belirtilerine sebep olduğu düşünülmektedir. Bağışıklık sistemi üzerinden etkileşime geçen bu parazit türü genelde; bağışıklık sisteminin düşük olması, bağışıklık sistemini baskılayan ilaçların kullanımı, kemoterapi tedavisi almak ve AIDS gibi bağışıklığın baskılanması durumlarında daha yüksek ihtimalle görülebilir.
Belirti ve Semptomlar
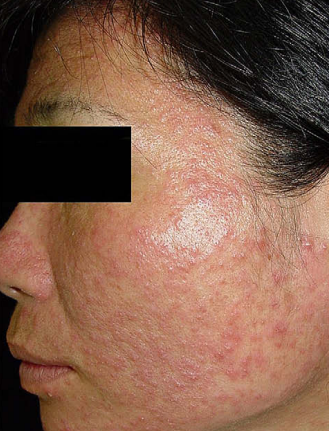
Demodikozis hastalığının en yaygın ve ilk belirtileri yüzde görünür. Ve şu belirtilere sebep olmaktadır;
- Yanma hissi
- Kaşıntı
- Pürüzlü yüzey
- Kızarık hassas cilt
- Beyaz folikülümsü görüntü
Aynı zamanda gözleri de etkileyen bu hastalık ise gözlerde şu belirtilere sebep olmaktadır;
- Gözde kaşıntı
- Gözde tahriş
- Görme duyusunda azalma
- Kirpik dökülmesi
- Pullu ve beyaz kalıntılar olan göz kapağı görüntüsü
Görülme Sıklığı
Demodikozis hastalağının speesifik bir görülme sıklığı olmadığı pek çok çalışma ile kanıtlanmıştır. Kadın ve erkek hastalar arasındaki kıyaslamaya bakıldığında da anlamlı bir ayrım bulunamamıştır. Ancak, yapılan bir araştırmada 16-96 yaş grubunda görülme sıklığı belirli aralıklarla değişse de genel oran %70’den fazla olarak belirtilmiştir.
Kalıtım Paterni/Deseni
Kalıtsal olmayan demodikozis hastalığı, parazit kaynaklı olmasından dolayı genetik yollarla aktarılamaz. Bu sebeple 15 yaş ve altı, özellikle yenidoğanlarda, bu parazite genelde rastlanmaz veya düşük orandadır. Ancak, zamanla insanlarla temastan dolayı ileri yaşta bu parazitlere hemen hemen herkeste rastlanmaktadır.
Teşhis Yöntemleri ve Tedaviler
Teşhis yöntemlerinin başında ise belirtilerin değerlendirilmesi sonrasında, dermateskop ile bölgeyi inceleme yöntemi gelmektedir. Dermateskop sayesinde parazitlere ait kalıntılar gözlemlenebilmektedir. Onun haricinde benzer bir yöntem olan, hastanın derisinden sürüntü örneği alma ve mikroskop altında analiz etme yöntemi de kullanılmaktadır.
Tedavi olarak ilaç tedavileri ve deri üstünden kimyasal kullanımı olmak üzere bir kaç çeşitli tedavi yöntemi bulunmaktadır. En yaygın olanı ise antibiyotik olan metronidazol kullanımıdır. Benzen heksaklorür olarak bilinen Lindan kullanımı da yaygındır. Ayrıca, bazı araştırmalarda da salisilik asit, selenyum sülfat ve potasyum hidroksit de tedavi amaçlı kullanılmış ve sonuç almıştır.
Hastalığın Diğer İsimleri
- Demodikidozis
- Demodikozis
- Demodeks
Referanslar
- Yazısız, H., Çekin, Y., & Koçlar, F. G. (2019). The Presence of Demodex Mites in Patients with Dermatologic Symptoms of the Face. Turkiye parazitolojii dergisi, 43(3), 143–148. https://doi.org/10.4274/tpd.galenos.2019.6062
- https://rarediseases.info.nih.gov/diseases/1802/demodicidosis
- https://www.orpha.net/
- Elston, C. A., & Elston, D. M. (2014). Demodex mites. Clinics in dermatology, 32(6), 739–743. https://doi.org/10.1016/j.clindermatol.2014.02.012
- https://www.consultant360.com/content/demodicidosis
- Hsu, C.-K., Hsu, M. M.-L., & Lee, J. Y.-Y. (2009). Demodicosis: A clinicopathological study. Journal of the American Academy of Dermatology, 60(3), 453–462. doi:10.1016/j.jaad.2008.10.058
- HUGUL, H., KECİCİ, A. S., & KUTLUBAY, Z. (2022). Treatment of Demodicosis with Potassium Hydroxide Dermabrasion. Practical Dermatology.
- Baima, B., & Sticherling, M. (2002). Demodicidosis revisited. Acta dermato-venereologica, 82(1), 3–6. https://doi.org/10.1080/000155502753600795

 [9]
[9]

{kind=link}
{kind=link}
{kind=link}
{kind=link}