Eş Acquired Hemophilia Hastalıkları:
• acquired hemophilia A (AHA) (Klasik hemofili)
• acquired hemophilia B (AHB) (Noel hastalığı)
Genel Bilgi
Kazanılmış (edinilmiş) hemofili (AH) kanama ile karakterize nadir görülen otoimmün bir hastalıktır. Otoimmün bozukluklar, vücudun bağışıklık sistemi yanlışlıkla sağlıklı hücrelere veya dokulara saldırdığında ortaya çıkar. AH’de vücut, en sık faktör VIII olan pıhtılaşma faktörlerine saldıran antikorlar (inhibitörler olarak bilinir) üretir. Pıhtılaşma faktörleri, kanın normal olarak pıhtılaşması için gereken özel proteinlerdir. Sonuç olarak, etkilenen bireyler, kaslara, cilde ve yumuşak dokuya, ameliyat sırasında veya travma sonrası anormal, kontrolsüz kanama ile ilişkili komplikasyonlar geliştirir. Spesifik semptomlar arasında burun kanaması (burun kanaması), vücutta morluklar, konjekte olmuş kanın (hematomlar) katı şişmesi, idrardaki kan (hematüri) ve gastrointestinal veya ürogenital kanamalar bulunur. AH, ağır vakalarda ciddi, hayatı tehdit edici kanama komplikasyonlarına neden olabilir. Hastaların yaklaşık% 50’sinde, tanımlanabilir bir altta yatan klinik durum vardır; diğer% 50’sinde ise hiçbir neden bilinmemektedir. AH, bazı pıhtılaşma faktörlerinin konjenital eksikliğinden kaynaklanan nadir bir genetik hastalık grubu olan konjenital hemofili’den farklıdır. Ana hemofili şekli X’i bağlayan bir hastalık olan hemofili A’dır (klasik hemofili), erkekleri de kadınları da etkileyebilir.
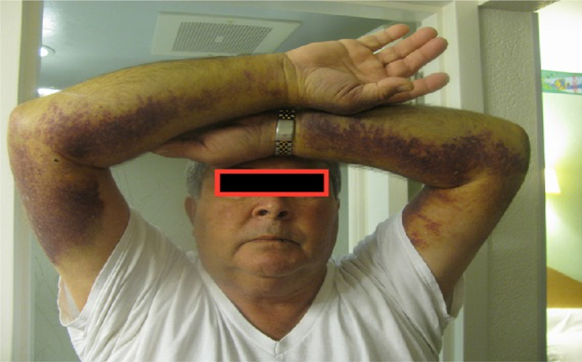
Görsel 1. Acquired Hemofili A ile ilgili kanama görüntüsü
kaynak: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4431493/#!po=14.0000
Faktörü VIII olmayan veya azalmış olan hastalarda konjenital hemofili A (“klasik” hemofili) ve IX olan veya olmayan faktörde IX olanlar konjenital hemofili B (Noel Hastalığı) olarak bilinir.
Genetik Değişiklikler / Etken Faktörler
Nedenleri
AH, otoimmün bir hastalıktır. Bağışıklık sistemi yanlışlıkla sağlıklı dokuya, özellikle de pıhtılaşma faktörü olarak bilinen özelleşmiş proteinlere, çoğunlukla pıhtılaşma faktörü VIII’e saldıran antikorlar ürettiğinde ortaya çıkar.
Bağışıklık sistemi normal olarak antikor adı verilen özel proteinler üreterek yabancı bir maddeye tepki verir. Antikorlar, yabancı maddeleri doğrudan yok ederek veya beyaz kan hücrelerinin yok etmesi için onları işaretleyen bir maddeyle kaplayarak çalışır. Antikorlar sağlıklı dokuları hedeflediklerinde, otoantikorlar olarak adlandırılabilirler. Araştırmacılar, tetikleyici bir olayın (bir enfeksiyon veya altta yatan bir bozukluk gibi) bağışıklık sistemini otoantikorlar üretmeye neden olabileceğine inanmaktadır. AH’deki otoantikorlar, inhibitör olarak adlandırılır çünkü etkilenen pıhtılaşma faktörünün fonksiyonunu inhibe ederler.
AH ağırlıklı olarak yaşlıların bir hastalığıdır. Hastaların yaklaşık% 50’sinde, altta yatan bir bozukluk veya tetikleyici olay tanımlanamaz (idiyopatik form). Kalan% 50, lupus, romatoid artrit, multipl skleroz, Sjögren sendromu ve temporal arterit gibi otoimmün rahatsızlıklar dahil olmak üzere birlikte varolan rahatsızlık veya rahatsızlıklara sahiptir; inflamatuar bağırsak hastalığı veya ülseratif kolit; enfeksiyonlar; şeker hastalığı; hepatit; solunum veya dermatolojik hastalıklar; kan (hematolojik) kanser veya bazı katı tümörler veya penisilin veya interferon gibi ilaçlarla ilişkisi. Gebelikle ilişki, esas olarak doğum sonrası dönemde bildirilmektedir.
Belirti vr Semptomlar
Hastaların yaklaşık 1 / 3’ünde kanamaları kontrol etmek için tedavi gerekmese de kanama şiddeti değişkendir ve hastaların 1 / 3’ünden fazlasında çoklu kanama atakları vardı. Deri altı kanaması (ekimozlar) AH’nin en sık görülen belirtisidir, bunu kas kanaması (hematom), gastrointestinal (melena), genitoüriner (hematüri) ve retroperitoneal izler. İntrakraniyal kanama nadirdir, ancak ölümcül olabilir. Konjenital hemofili A’nın aksine eklem kanaması nadirdir.
Kanama genellikle sebepsiz olarak oluşur (kendiliğinden). Kanama bölümleri genellikle ağırdır ve hayatı tehdit edici olabilir. Bazı hastalarda, gecikmiş tanı ve ek tıbbi sorunların varlığı genellikle genel şiddetine etki eden faktörlerdir. Yumuşak dokulara kanama hızlı bir şekilde ilerleyebilir, potansiyel olarak kaslar, sinirler ve Bu yapıların sıkışması nedeniyle en sık olarak kol ve bacaklarda bulunan kan damarları etkilenir.
Etkilenen Bireyler ayrıca, ameliyat sırasında veya travma sonrası, hatta önemsiz olsa bile, aşırı kanama riski altındadır. Gebe kadınlarda genital ağır kanama, özellikle doğum sonrası (doğum sonrası dönem) ortaya çıkabilir.
Genetik Görülme Sıklığı
AH, önceden eşit kanama bozukluğu öyküsü olmayan ve yaklaşık olarak eşit sayıda erkek ve kadın etkilenen bireylerde gelişir. Amerika Birleşik Devletleri’nde, bozukluğun genel popülasyonda yılda 1.000.000 / yılda yaklaşık 0.2-1 kişiyi etkilediği tahmin edilmektedir. Birleşik Krallık’ta, hastalığın 1.000.000 / yılda 1.4’ü etkilediği tahmin edilmektedir. Ancak, etkilenen bireyler teşhis edilmeyebilir veya yanlış teşhis edilebilir, bu da hastalığın genel popülasyondaki gerçek sıklığını belirlemeyi zorlaştırır. Vakaların çoğu faktör VIII (AHA) eksikliğini içerir. Faktör IX (AHB) eksikliğini içeren vaka daha az sayıda tanımlanmıştır.
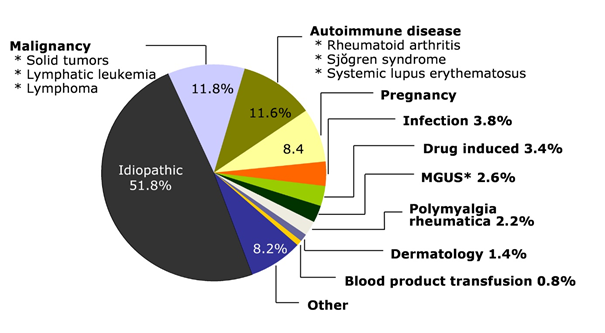
Görsel 2. Edinilmiş Hemofili ile İlişkili Koşullar
kaynak: https://www.rarebleedingdisorders.com/bleeding-disorders/acquired-hemophilia.html
Kalıtım Paterni/Deseni
Hemofili vakalarının çoğunluğu genetiktir, erkekleri etkiler ve çocukluk çağında oldukça erken teşhis edilir. Ancak, daha sonra yaşamda gelişen ve “kazanılmış hemofili” olarak adlandırılan nadir görülen hemofili vakaları vardır.
Edinilmiş hemofili, hastaların milyonda birini etkileyen çok nadir görülen bir durumdur. Genellikle hastalara uzun süreli kanama olduğunda veya kaza v.s. sebeplerle travma sonrası, bacaklarda ve kollarda olağan yaralanma ile ilgili morarmalardan çok farklı görünen geniş morarmalarda teşhisi konur. Basit bir kan testi, pıhtılaşma faktörü VIII eksikliği olup olmadığını belirleyebilir. Eğer tedavi edilmezse, alınan hemofili çok ciddi ve yaşamı tehdit edici olabilir. Bununla birlikte, immünosüpresif ilaçlar ve pıhtılaşma faktörü enjeksiyonları ile teşhis konduktan sonra kolayca tedavi edilir.
Bu hastalıkta kadınlar taşıyıcıdır. Taşıyıcı bir annenin erkek çocuğuna %50 oranında hemofili hastalığını geçirme olasılığı vardır. Hemofili A (faktör VIII eksikliği), yaklaşık her 5.000 erkek doğumunda bir ortaya çıkar ve hemofili olgularının yaklaşık %80 – %85’ini oluşturur. Hemofili B (faktör IX eksikliği) daha nadir olup her 30.000 erkek doğumunda bir görülür.
Teşhis Yöntemi ve Tedavi
Teşhis
AH klinik tablodan şüphelenmeli ve anormal bir pıhtılaşma testi ile doğrulanmalıdır. Son zamanlarda anormal kanaması olan ve aktive parsiyel tromboplastin zamanı (aPTT), özellikle yaşlı ve peri ve post-postum kadınlarda izole uzun süreli olan hastalarda tanı düşünülmelidir.
Klinik Test ve Çalışma
Rutin birinci satır pıhtılaşma testleri arasında aktive parsiyel tromboplastin zamanı (aPTT) ve protrombin zamanı (PT) bulunur. İki test, iki farklı doku faktörü (aPTT parsiyel tromboplastinde) ile tetiklenen plazmanın koagülasyon süresini ölçer. aPTT, temel olarak FVIII, FIX, FXI ve XII’ye duyarlıdır; PT, karaciğer tarafından sentezlenen pıhtılaşma proteinlerine duyarlıdır (FII, FVII, FIX, “protrombin kompleksi” olarak adlandırılır; K vitaminine bağlı sentez ile).
AH’li bireylerde, normal PT ile izole edilmiş bir uzun süreli aPTT vardır. Spesifik olmayan inhibitörler (örneğin, lupus antikoagülan) veya heparin tedavisi gibi izole uzun süreli aPTT’nin diğer nedenlerini ekarte etmek için testler de yapılır.
Hastanın plazmasını normal plazma ile karıştırmak suretiyle gerçekleştirilen aPTT karıştırma testleri, teşhisi daha da doğrulayabilir. Bir karıştırma çalışması, genetik faktör eksikliklerini faktör inhibitörlerinden ayırmaktadır. Bir kan numunesi alınır ve kontrol grubundan alınan kanla karıştırılır. Faktör eksikliği olan bireylerde normal plazma test değerini normale döndürür; Faktör inhibitörü olan bireylerde yok.
Bir faktör inhibitörü kurulduktan sonra, koagülasyon faktörlerinin (çoğu durumda FVIII) aktivitesini ve inhibitörün titresini ölçmek için bir analiz yapılacaktır. AHA’lı bireylerde bu faktör VIII eksikliği gösterecek ve ciddiyeti de belirleyecektir.
Tedavi
AH nadir görülen bir hastalık olduğundan, etkilenen bireyleri tedavi etmek için kullanılan çoğu tedavi raporlarına veya küçük vakalara dayanmaktadır. Spesifik tedavilerin birbirleriyle etkinliklerini karşılaştıran az sayıda çalışma vardır. Sonuç olarak, tedavi oldukça kişiselleştirilmiştir.
Spesifik terapötik prosedürler ve müdahaleler, mevcut spesifik semptomları içeren sayısız faktöre bağlı olarak değişecektir; altta yatan neden de dahil olmak üzere hastalığın doğal seyri (biliniyorsa); yaş ve genel sağlık (örneğin eşlik eden hastalık), bazı ilaçların veya prosedürlerin toleransı ve kişisel tercih; ve diğer faktörler. Özel terapötik müdahalelerin kullanımına ilişkin kararlar, doktorun ve sağlık ekibinin diğer üyeleri tarafından, vakasının özelliklerine dayanarak, hasta ve / veya ebeveynleri ile dikkatli istişare içinde verilmelidir; olası yan etkiler ve uzun vadeli etkiler dahil olmak üzere potansiyel fayda ve risklerin kapsamlı bir tartışması; hasta tercihi; ve diğer uygun faktörler.
Spontan remisyon bildirildi; genel olarak doğum sonrası durumlarda (doğumdan sonraki birkaç ay içinde) ve alerjik ilaç reaksiyonuna ikincil durumlarda, genellikle rahatsız edici ilacı bıraktıktan birkaç ay sonra ortaya çıkabilir. Spontan remisyon, düşük titre inhibitörleri olanlar gibi etkilenen diğer bireylerde de görülebilir.
AH’nin yönetimi aşağıdaki hedeflere odaklanır: kanamanın kontrol edilmesi ve önlenmesi (varsa veya önemliyse), inhibitörün ortadan kaldırılması ve altta yatan hastalığın tedavisi (varsa).
Kanama Bölümlerini Kontrol Etme
Kanama çok şiddetli olabilir ve ani bir başlangıç olabilir. Bu nedenle morbidite ve mortaliteyi azaltmak için hızlı hemostatik kontrol zorunludur. Uluslararası Tavsiyeler, anti-hemorajik tedavinin, inhibitör titresi ve faktör VIII aktivitesinden bağımsız olarak AH tanısının doğrulandığı ciddi kanaması olan hastalarda başlatılması gerektiğini belirtir. İki yaklaşım vardır: bypass edici ajanların kullanımı (kazanılmış eksikliği atlayan faktörlerin konsantreleri) veya FVIII seviyelerini arttırma stratejilerinin kullanılması. Bu iki seçenek arasındaki seçim, bölgeye ve kanamanın ciddiyetine ve her bir hastanın özelliklerine dayanır. Hemostatik ajanlar öngörülebilir bir etkiye sahip olmadığından, kanama tedavisi bu alanda uzman bir kişi tarafından denetlenmeli ve hemostatik kontrol ve kanama çözünürlüğünü doğrulamak için uygun laboratuvar testleri, görüntüleme teknikleri ve yetenekli bir klinik değerlendirme gereklidir. Fibrin yapıştırıcı veya antifibrinolitik ajanlar, yerel kanamanın kontrolünde faydalı olabilir. Bypassing ajanları, hızlı hareketleri ve yüksek etkinlik seviyeleri nedeniyle önerilen birinci basamak tedavidir. Dozaj büyük ölçüde konjenital hemofili hastalarında FVIII inhibitörleri olan hastaların yönetimi konusundaki deneyime dayanır ve genellikle klinik değerlendirmeye dayanır.
Halen mevcut olan bypass yapan ajanlar, rekombinant aktif faktör VII (rFVIIa veya NovoSeven® RT) veya aktifleştirilmiş protrombin kompleks konsantresidir (aPCC veya FEIBA®). Bu tedavilerin hiçbiri tüm bireylerde etkili değildir.
NovoSeven® RT, faktör VII’nin genetik olarak tasarlanmış (rekombinant) bir versiyonudur. Yapay olarak bir laboratuvarda üretildiği için, insan kanı veya plazması içermez ve sonuç olarak, kan kaynaklı virüsler veya bu gibi diğer patojenlerin riski yoktur. NovoSeven iyi tolere edildi ve çok az yan etkiyle ilişkilendirildi. AH’li bireylerde trombotik yan etki riski (tromboz)% 1’in altındadır. NovoSeven, edinilmiş hemofili hastalarının tedavisinde baypas edici ajan olarak kullanılmak üzere Gıda ve İlaç İdaresi (FDA) tarafından onaylanmıştır.
aPCC, çeşitli aktif pıhtılaşma faktörlerini içeren plazma kaynaklı, anti-inhibitör kompleksidir. Bu faktörler, ilacın kan pıhtılarının oluşumundaki bazı faktörleri atlamasına izin verir (faktör VIII gerektiren basamaklar dahil). aPCC, olası virüsleri veya benzer patojenleri etkisiz hale getirmek için tedavi edilir ve ters trombotik olaylar nadirdir. Şu anda Amerika Birleşik Devletleri’nde mevcut olan aPCC’nin tek şekli, şu anda Shire’ın bir parçası olan Baxalta’dan temin edilebilen FEIBA®’dır. Daha fazla bilgi için iletişime geçin: Baxalta http://www.feiba.com/
FVIII’in, endotel hücreleri tarafından FVIII salınımını indükleyen, FVIII konsantresi veya DDAVP infüzyonu gibi bir FVIII artışına izin veren terapötik yöntemler, inhibitör titresi çok düşük olmadığı sürece (yani <5 Bethesda üniteleri [BU)) ve genellikle yetersiz olarak kabul edilir. atlayan ajanlar mevcut değildir. 2014 yılında ABD FDA, edinilmiş hemofili A (edinilmiş Faktör VIII [FVIII] eksikliği olan yetişkinlerde kanama ataklarının tedavisi için Obizur’ü onayladı. Obizur, şimdi Shire’ın bir parçası olan Baxalta tarafından üretildi. Daha fazla bilgi için: Baxalta http://www.obizur.com/
İnhibitör Eradikasyonu
Bazı durumlarda inhibitör, inhibitör bulunduğu sürece kendiliğinden kaybolabilse de, kanamayla ilişkili morbidite ve mortalite önemlidir. Bu nedenle, yetişkinlerde inhibitörün yok edilmesi (iimunosupresyon tedavisi) tedavisinin, açıkça kontrendike olmadıkça AH tanısından hemen sonra başlaması tavsiye edilir. Öneriler, büyük ölçüde, gerçek hayat verilerini toplayan kayıt defterlerinde yapılan gözlemlerden elde edilir.
Genel olarak, tek başına veya siklofosfamid ile ilişkili kortikosteroidler birinci basamak tedavidir. Bu iki yöntemde uzun süreli sağkalımda belirgin bir fark gözlenmedi. Bununla birlikte, bireyler immünosüpresif ilaçlara farklı tepkiler verir ve bir bireyde etkili olan diğerinde etkili olmayabilir. Siklosporin A, azatiyoprin, vinkristin, mikofenolat mofetil ve 2-klorodeoksiadenozin de dahil olmak üzere edinilmiş hemofili tedavisi için çeşitli ek immünosüpresif maddeler kullanılmıştır.
Tedaviye cevap kriterleri henüz belirlenmemiştir; Bununla birlikte, yüksek inhibitör titresi ve düşük FVIII düzeyi tedaviye yanıtı öngörüyor gibi görünmektedir.
AH’nin nüksetmesi, immünosüpresif tedavi durdurulduktan sonra veya doz azaltıldığında remisyon sağlayan kişilerde ortaya çıkabilir. İlişkili yan etkiler nedeniyle, uzun süreli immünsüpresif tedavi önerilmemektedir.
AH’li bireylerin, inhibitör eradikasyonundan sonraya kadar önemli bir travma riski taşıyan etkinliklerden kaçınmaları teşvik edilir.
AH’li hastalar federal olarak finanse edilen hemofili tedavi merkezine yönlendirmeden faydalanacaktır. Bu ihtisas merkezleri hemofili hastalığına ve spesifik tedavi planlarının geliştirilmesi, etkilenen bireylerin izlenmesi ve takibi ve son teknoloji tıbbi bakım da dahil olmak üzere ilgili hastalıklar için kapsamlı bakım sağlayabilir. Bir hemofili tedavi merkezinde tedavi, bireylerin ve aile üyelerinin, hemofili hastalarının tedavisinde deneyimli profesyonel bir sağlık ekibi (doktorlar, hemşireler, fizyoterapistler, sosyal hizmet uzmanları ve genetik danışmanlar) tarafından korunmalarını sağlar.
Araştırma Terapileri
Araştırmacılar, AH’li bireyler için potansiyel bir terapi olarak, rituximab ilacını inceliyorlar. Bu ilaç, hastalığa neden olan otoantikorlara saldırır. Rituximab, monoklonal bir antikor veya biyolojik terapi olarak sınıflandırılır – antikorlar gibi hareket eden ilaçlar, ancak yapay olarak bir laboratuvarda yaratılırlar. Başlangıçta, rituximab inhibitörlerin yok edilmesinde ümit verici sonuçlar göstermiştir. Bazı araştırmacılar, kortikosteroid / siklofosfamid tedavisine cevap vermeyen bireylerde inhibitörlerin yok edilmesi için ikinci basamak bir tedavi olarak veya kortikosteroid / siklofosfamid tedavisini tolere edemeyen bireylerde birinci basamak tedavi olarak rituximab önermişlerdir. AH tedavisi için rituksimabın uzun vadeli emniyetini ve etkinliğini belirlemek için daha fazla araştırma gereklidir.
AH’de inhibitörleri yok etmek için yüksek dozda intravenöz immünoglobulin kullanılmıştır. Bununla birlikte, tıbbi literatürdeki çoğu rapor hayal kırıklığı yaratan sonuçları detaylandırmaktadır. Bazı araştırmacılar, bu tedavinin, inhibitörleri yok eden diğer ilaçlara veya prosedürlere yardımcı bir tedavi olarak en iyi şekilde söz verdiğine inanmaktadır.
Yüksek inhibitör titresi ve şiddetli kanamaya sahip bazı kişiler, plazmaferez veya immüno-emilim adı verilen bir prosedüre maruz kalabilir. Bu prosedürler genellikle diğer tedavi seçeneklerine cevap vermeyen ve hayatı tehdit edici kanama bölümleri yaşayan hastalar için ayrılmıştır. Plazmaferez, istenmeyen maddelerin kandan uzaklaştırılmasını içerir. Kan hastadan çıkarılır ve katı kan hücreleri sıvı plazmadan ayrılır. Hastanın plazması daha sonra, orijinal kan hücreleri ile birlikte yeniden transfekte edilen donör insan plazması veya albümini ile değiştirilir. Modifiye Bonn-Malmö Protokolü (MBMP), immünoglobulinleri (FVIII inhibitörü) FVIII replasmanı ve immünosüpresyonu ile spesifik olarak bağlayan kolonlarda immü emilimini birleştirir ve akut kanamanın hızlı ve güvenli kontrolünü sağlayabilir. Esas olarak Avrupa’da uygulanmaktadır.
İmmünsüpresif tedavinin komplikasyonları (IST)
IST zaten yaşlı, kırılgan popülasyonda enfeksiyon riskini arttırır. Nötropeni (beyaz kan hücrelerinin azalması) ve sepsis sıklıkla bildirilir ve ölüme katkıda bulunur. Yükseltilmiş kan şekeri düzeyleri, gastrointestinal ülserler, kas bozuklukları ve psikiyatrik bozukluklar (% 3) dahil olmak üzere diğer iyi bilinen steroid tedavisi komplikasyonları bildirilmiştir.
Sonuçlar, Araştırma ve Diğer İhtiyaçlar
AH, hastayı değerlendiren ilk kişi olan internist veya acil servis doktoru tarafından daha sık karşılaşılır. AH’nin gerçek dünyadaki klinik uygulamalarda tanı konmaması ve yanlış tanı konması muhtemeldir, bu durumun sağlık hizmetleri pratisyenleri arasında bu hastalığa ilişkin farkındalık yaratması ve AH’nin yönetimindeki uzmanlara yönlendirmeyi teşvik etmesi gerektiğini gösterir. Birçoğu için, AH, derhal tanınması ve uygun yönetimin net faydaları ile büyük ölçüde tedavi edilebilir. Hemostatik hem de immün tedavi, önemli riskler getirir ve güvenliği sağlamak için yakın izleme gerektirir. AH araştırmalarında en yüksek öncelik, tedavinin yan etkileri ile daha iyi dengeleyici inhibitör eradikasyonu ile sağkalımı artıran daha güvenli immünsüpresif rejimler geliştirmektir.
Hastalıkla İlişkili Genler
İlgili Bozukluklar
Aşağıdaki bozuklukların belirtileri AH’ninkine benzer olabilir. Karşılaştırmalar ayırıcı tanı için faydalı olabilir. Hemofili, bir grup nadir kanama bozukluğu grubu için genel bir terimdir. Hemofili hastalarının çoğu, inaktif veya eksik kan proteinlerinin neden olduğu kan pıhtılaşması (pıhtılaşma) bozukluğudur. Üç ana kalıtsal hemofili şekli vardır: hemofili A (aynı zamanda klasik hemofili, faktör VIII eksikliği veya antihemofilik globulin [AHG] eksikliği olarak da bilinir); hemofili B (Noel hastalığı veya faktör IX eksikliği); ve hemofili C (faktör XI eksikliği). Hemofili A ve B, X’e bağlı resesif genetik bozukluklar olarak kalırken, hemofili C, otozomal resesif genetik hastalık olarak kalıtılır. Hemofili A ve B çoğunlukla erkeklerde ifade edilir, ancak kadınlar da etkilenebilir. Hemofili C erkekleri ve dişileri eşit sayıda etkiler. Hemofili hafif, orta veya şiddetli olarak sınıflandırılabilir; şiddeti seviyesi kandaki aktif pıhtılaşma faktörü yüzdesiyle belirlenir (normal yüzde 50 ila 150 arasında değişmektedir). Şiddetli hemofili hastaları kanlarında aktif pıhtılaşma faktörünün yüzde birinden daha azına sahiptir. (Bu hastalıklar hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “hemofili” yi seçin.)
Von Willebrand hastalığı (VWD), uzun süreli kanama ile sonuçlanan kalıtsal bir kanama bozukluğudur ve etkileri bakımından geniş ölçüde değişkenlik gösterir. VWD’li bireylerde von Willebrand faktörü eksikliği veya eksikliği vardır. Ayrıca, VIII faktörü olarak bilinen düşük bir ilave faktör seviyelerine sahip olabilirler. Willebrand faktörü eksik veya kusurlu, trombositlerin yanlış işleyişi, hemostazın erken dönemlerinde yer alan özel kan hücresi fragmanları, kanamayı durdurmak için damar duvarı lezyonu bölgesinde tıkaç oluşumu ile sonuçlanır. VWD’li bireylerde trombositler kan damarlarındaki deliklere yapışmaz ve kanama uzar. Çoğu insan nispeten hafif bir hastalığa sahiptir ve yetişkin olana kadar teşhis edilmez. Bireylerin küçük bir yüzdesi, bebeklik döneminde veya erken çocukluk döneminde, uzun süreli kanama ve anormal derecede yavaş bir pıhtılaşma süresi gibi problemler yaşamaya başlar. Belirtileri gastrointestinal kanama, burun kanamaları, diş eti kanaması ve kolay morarma olabilir. Etkilenen kişiler, yaralanma, doğum ve / veya ameliyattan sonra kolayca kanabilir. Hastalığın üç ana formu vardır. Otozomal dominant bozukluk olarak iki tip kalıtımsaldır; üçüncü tip otozomal resesif bir hastalık olarak kalıtsaldır. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “von Willebrand” ı seçin.)
Hastalığın Diğer İsimleri
⁃ Kazanılmış (Edinilmiş) Hemofili
⁃ Edinsel Hemofili
⁃ AH
⁃ AHA – AHB
Kaynaklar
⁃ https://rarediseases.org/rare-diseases/acquired-hemophilia/
⁃ https://hemophilianewstoday.com/2017/06/13/understanding-acquired-hemophilia/?amp
⁃ https://www.rarebleedingdisorders.com/bleeding-disorders/acquired-hemophilia.html
⁃ http://www.novonordisk.com.tr/hastalara-ozel/hemostaz/konjenital-hemofili.html