Genel Bilgi
Ensefalosel (sefalosel, kranial veya oksipital meningosel, kranium bifidum, ensefalomeningosel, ensefalomeningositosel), beynin kese şeklindeki protrusyonlar ve onları örten zarlar tarafından karakterize edilmiş nadir nöral tüp defektleridir. Kafatasındaki yapılar, bir kusurdan dışarı taşar Fıtıklaşmış kesede menenj ve beyin dokusu (ensefalosel) veya sadece menenj (meningosel) bulunabilir. Bu defektler, fetüs gelişimi esnasında nöral tüplerin tamamen kapanmamasında dolayı olur. Bunun sonucunda, kafatasının orta üst kısmında, alın ve burun arası alanda ya da kafatasının arkasında aşağı yönde bir oluk oluşur. Kafatasının arkasında konumlanan bu oluk, ensefaloseller nörolojik problemlerle ilişkilendirilir. Ensefaloseller, çoğunlukla doğumdan hemen sonra tanısı konulan dramatik deformelerdir; ancak nazal ve alın bölgelerinde oluşan küçük ensefaloseller, sıklıkla tespit edilemez. Bu hastalığın genetik bir bileşeni vardır; diğer aile üyelerinin geçmişinde spina bifida denilen açık omurga hastalığı ya da anensefali hastalığı görülen insanlarda, bu hastalık daha sık ortaya çıkar. [6]
Ensefalosel anormalliğinin farklı tipleri vardır. Oksipital ensefalosel olarak adlandırılan knobloch sendromu, kafatasında kese şeklinde protrusyonların, oksipital kemiğinde defekte olmasıdır. Ensefaloseller, zihinsel engellilikle ilişkilendirilebilir; fakat knobloch sendromuna sahip çoğu hastada zihinsel engellilik gözlemlenmemiştir. Robert sendromu olarak adlandırılan bu tipte, alın kısmında çıkıntı oluşumu görülür. Robert sendromu görülen bireylerde doğum öncesi ve doğum sonrası yavaş büyüme görülür. Bu sendroma sahip bireylerde damak yarıklığı, küçük çene, hipertelorizm denilen gözler arası açıklığı ve benzeri birçok anormallikler görülür. Buna ek olarak, bu sendroma sahip bireylerde kalp, böbrek ya da genital bölgelerinde anormallikler görülebilir.
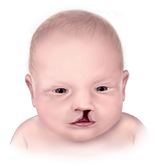
Figür 1: Dudak yarıklığı örneği.[2]
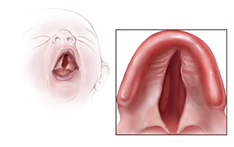
Figür 2: Damak yarıklığı örneği. [2]
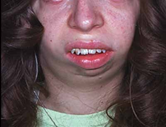
Figür 3: Küçük çene örneği. [2]
Genetik Değişiklikler ve Etken Faktörler
ESCO2 geninde oluşan mutasyon, Robert sendromuna sebep olur. Bu gen, hücre bölünmesi esnasında uygun kromozom ayrımını yapmayı sağlayan proteinin kodlanmasında görev alır. Hücreler bölünmeden önce tüm kromozomlarını kopyalarlar. Her bir kromozomdaki DNA, kardeş kromatidler denilen ikiz yapılar şeklinde sıralanır. Bu olayda, ESCO2 proteini kardeş kromatidleri, hücre bölünmesi esnasına kadar bir yapışkan gibi bir arada tutar. [2]
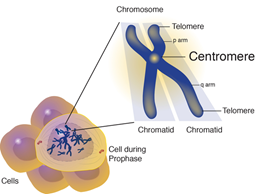
Figür 4: Kromozomun Şematik gösterimi.

Figür 5: Oklarla gösterilmiş kısımlar, sentromerler.
ESCO2 genindeki tanımlanmış tüm mutasyonlar, hücrenin fonksiyonel ESCO2 projenin üretmesini engelliyor. Bu da, bazı kardeş kromatidler arasındaki o yapışkan bağların kaybına neden olur. Roberts sendromunda hücreler, hücre bölünmesini geciktirerek anormal kardeş kromatid bağlanmasına tepki verirler. Bu durumda gecikmiş hücre bölünmesi, hücreye “kendini yok etmelisin” sinyali verir. Robert sendromunun semptom ve belirtileri, erken gelişme esnasında çeşitli dokulardan hücre kaybını sonuçlandırabilir. Hafif ya da ağır olarak iki türlü de etkilenmiş bireylerde fonksiyonel ESCO2 proteini bulunmadığından, bu değişen şiddetin altında yatan neden tam olarak anlaşılmış değil. Araştırmacılar, diğer genler ve çevresel etkenlerin de buna dahil olduğunu söylüyorlar.
Genetik Görülme Sıklığı
Ensefalosel, tüm kranyospinal disfarizmaların %10 ila %20’sini oluşturmaktadır. Ensefalosel, yaklaşık 0.8:10,000 ila 4:10,000 arasında canlı doğumlarda ve Güneydoğu Asya’da daha yüksektir (2:10,000). Genel olarak, arka ansefaloseller, anterior efektlerden daha sık görülür. [4]
Kalıtım Paterni
Kalıtım paterni göz önünde bulundurulduğunda bu kondisyon, otozomal resesif paternde kalıtsaldır. Bu da demek oluyor ki her hücrede bulunan genin, her iki kopyasıda mutanttır. Otozomal resesif kondisyona sahip bir bireyin ebeveynleri, bu mutant genin bir kopyasını taşırlar, fakat fenotiplerinde semptomları ya da belirtilerini göstermezler.[2]
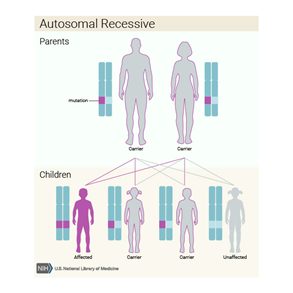
Figür 5: Ensefalosel’in kalıtım paterni. [5]
Teşhis Yöntemleri ve Tedavileri
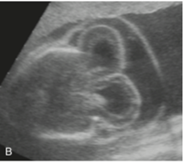
Figür 6: Ensefalosel sendromlu bir fetüs.[4]
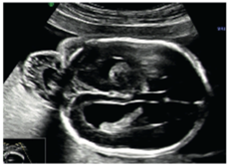
Figür 7: 24. Haftada ensefalosel.[4]
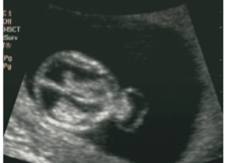
Figür 8: 12. Haftada ensefalosel.
Bu hastalığın teşhisinde, MRI cihazı oldukça kullanışlı bir alettir. Embryonik gelişme esnasında, MRI cihazı ile bebeğin kafatasındaki anormallikler kolaylıkla incelenebilir. Pariyetal ve frontal ensefaloseller, Asya’nın belli bölgelerinde daha çok yaygındır. Fetüslerde ensefaloselin öngörülebilirliği düşüktür. Hayatta kalma oranları azalmış ve zeka geriliğinin yaygınlığı kayda değerdir. Ensefaloselin önceden anlaşılabilmesi defekti büyüklüğüne, fıtıklaşmış beyin dokusunun varlığına ve bağlantılı anormallik ve sendromlara dayanır.
Bu hastalığın tedavi yöntemleri göz önüne alındığında, ensefalosel hastalığına sahip çocuklar için sıklıkla cerrahi müdahale zorunlu oluyor. Bu cerrahi yöntem, doğum anı ile 4 yaş arasındaki bireylerde uygulanabilir ama bu cerrahi yöntemin yapılabilmesi sendromun boyutuna, konumuna ve bağlantılı diğer komplikasyonlara aynı zamanda bir deri tabakasının ensefaloseli kaplayıp kaplamadığına bağlıdır. Eğer ensefaloselin üzerinde bir deri tabakası varsa ve bu koruyucu özelliği gösteriyorsa, cerrahi müdahale birkaç ay uzatılabilir. Eğer ensefaloselin üzerinde herhangi bir deri tabakası bulunmuyorsa, doğumdan sonra hemen cerrahi operasyona alınması önerilir.
Bu cerrahi müdahale, ensefaloselin dışarı çıkmış içeriği tekrar kafatasına koyma ile gerçekleşir. Beyin cerrahı, kafatasının belli bir bölgesini keserek çıkarır ve beyne ulaşmayı mümkün kılar. Daha sonra, dura mater denilen beynin üstünü örten sert dış katmana doğru keser.
Cerrah daha sonra beynin fıtıklaşmış parçasını, meninksleri ve sıvıyı tekrar kafatası içine koya ve çevreleyen keseyi uzaklaştırır. Daha sonra, dura mater kapatılır ve ve kafatasını ya başta çıkarttıkları parçayı takarak, ya da yapay bir malzeme kullanarak onarırlar. Bir ensefaloselin cerrahi operasyonla düzeltilmesinde, büyük ensefalosellerde bile, başka bir fonksiyonel sakatlığa neden olmadan sağlanabilir.
Bazı araştırmalar gösterdi ki, hamile olma ihtimali olan kadınların beslenmelerine ekleyecekleri folik asit ( bir tür B vitamini), nöral tüp defektleri riskini düşürmektedir. [3]
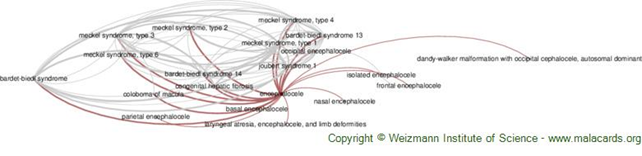
Hastalıkla ilişkili Genler
-Bulunan en bağlantılı genler[1]
-CEP290
-COL18A1
-MKS1
-TMEM67
-CC2D2A
Hastalığın Diğer İsimleri [2]
- Appelt-Gerken-Lenz sendromu
- Hipomelia hipotrikoz yüz hemanjiyom sendromu
- Psödothalidomid sendromu
- RBS
- Roberts-SC phocomelia sendromu
- SC phocomelia sendromu
- SC psödothalidomid sendromu
- SC sendromu
- Tetraphocomelia-yarık damak sendromu
Kaynaklar
[1]- https://www.malacards.org/card/encephalocele
[2]– https://ghr.nlm.nih.gov/condition/roberts-syndrome#statistics
[3]- https://rarediseases.org/rare-diseases/encephalocele/
[4]- T. Ghi, A. Dall’asta, G. Pilu, E. Contro, F. De Musso, T. Frusca – Obstetric Imaging: Fetal Diagnosis and Care: Second Edition- 2017- 213-226.e2
[5]- https://ghr.nlm.nih.gov/gallery
[6]- https://rarediseases.info.nih.gov/diseases/6333/encephalocele