Hastalığın Diğer İsimleri
- Yapısal karaciğer fonksiyon bozukluğu
- Ailevi hemolitik olmayan sarılık
- Gilbert hastalığı
- Gilbert-Lereboullet sendromu
- Gilbert hastalığı
- Gilbert sendromu
- Hiperbilirubinemi 1
- Meulengracht sendromu
- Konjuge olmayan benign bilirubinemi
Genel Tanı
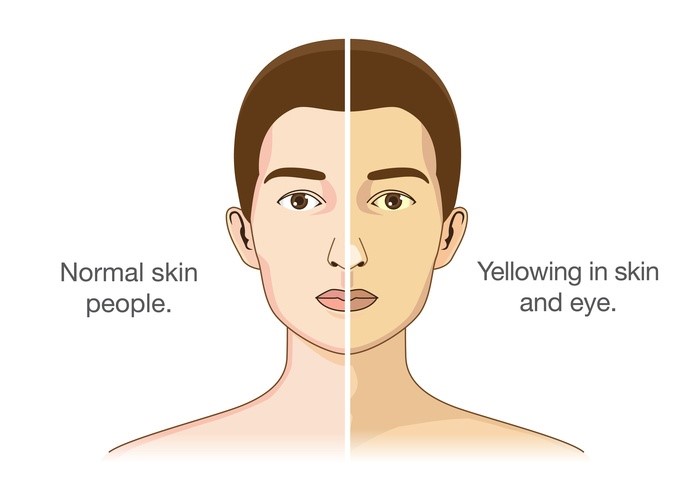
Gilbert sendromu, kandaki bilirubin (hiperbilirubinemi) adı verilen toksik bir maddenin yüksek seviyelerindeki periyotlarla karakterize edilen nispeten hafif bir durumdur. Turuncu kan rengine sahip olan bilirubin, kırmızı kan hücreleri parçalandığında üretilir. Bu madde vücuttan ancak karaciğerde kimyasal bir reaksiyona girdikten sonra çıkarılır; bu, bilirubinin (konjuge olmayan bilirubinin) toksik formunu konjuge bilirubin adı verilen toksik olmayan bir forma dönüştürür. Gilbert sendromlu kişilerde kanlarında konjuge olmayan bilirubin (konjuge olmayan hiperbilirubinemi) vardır. Etkilenen bireylerde, bilirubin seviyeleri değişkendir ve cildin ve gözlerin beyazlarının sararması olan sarılık yaratan seviyelere çok nadir olarak yükselir.
Gilbert sendromu genellikle ergenlikte tanınır. Bu rahatsızlığı olan kişilerde hiperbilirubinemi epizotları varsa, bu epizotlar genellikle hafiftir ve tipik olarak vücut stres altındayken, örneğin dehidrasyon, yiyeceksiz uzun süreli süreler, hastalık, kuvvetli egzersiz veya adet görme nedeniyle ortaya çıkar. Gilbert sendromlu bazı kişilerde abdominal rahatsızlık veya yorgunluk görülür. Bununla birlikte, Gilbert sendromlu kişilerin yaklaşık yüzde 30’unda durumun hiçbir belirtisi veya belirtisi yoktur ve sadece rutin kan testleri konjuge olmayan bilirubin seviyelerinde artış gösterdiğinde keşfedilir.
Oluşum Nedeni
UGT1A1 genindeki değişiklikler Gilbert sendromuna neden olur. Bu gen, öncelikle karaciğer hücrelerinde bulunan ve bilirubinin vücuttan çıkarılması için gerekli olan, bilirubin idrar difosfat glukuronosiltransferaz (bilirubin-UGT) enzimini yapmak için talimatlar sağlar.
Bilirubin-UGT enzimi, glukuronidasyon adı verilen kimyasal bir reaksiyon gerçekleştirir. Bu reaksiyon sırasında, enzim, konjüge edilmiş bilirubine dönüştürerek, konjüge edilmemiş bilirubine glukuronik asit denilen bir bileşiği aktarır. Glukuronidasyon, bilirubinin suda çözünebilir olmasını sağlar, böylece vücuttan çıkarılabilir. Gilbert sendromu dünya çapında görülür, ancak bazı mutasyonlar belirli popülasyonlarda daha sık görülür. Birçok popülasyonda, Gilbert sendromuna neden olan en yaygın genetik değişiklik (UGT1A1 * 28 olarak bilinir), bilirubin-UGT enziminin üretimini kontrol eden, promotör bölgesi adı verilen UGT1A1 geninin yakınındaki bir alanda meydana gelir. Bu genetik değişim enzim üretimini bozar. Bununla birlikte, bu değişiklik Asya popülasyonlarında nadir görülür ve etkilenen Asyalılar genellikle bilirubin-UGT enziminde tek bir protein yapı bloğunu (amino asit) değiştiren bir mutasyona sahiptir. Hatalı mutasyon olarak bilinen bu tip mutasyon, enzim fonksiyonunun azalmasına neden olur.
Gilbert sendromlu kişiler normal bilirubin-UGT enzim fonksiyonunun yaklaşık yüzde 30’una sahiptir. Sonuç olarak, konjuge olmayan bilirubin yeterince hızlı bir şekilde glukuronize edilmez. Bu toksik madde vücutta birikir ve hafif hiperbilirubinemiye neden olur.
Gilbert sendromuna neden olan genetik değişiklikleri olan herkes, glukuronidasyon sürecini daha da engelleyen koşullar gibi ek faktörlerin, durumun gelişimi için gerekli olabileceğini belirten hiperbilirubinemi geliştirmez. Örneğin, kırmızı kan hücreleri çok kolay bir şekilde parçalanabilir ve bozulmuş enzimin dayanamayacağı fazla miktarda bilirubin serbest bırakır. Alternatif olarak, bilirubinin glukuronidasyonun yapılacağı karaciğere hareketi bozulabilir. Bu diğer faktörler, diğer genlerdeki değişikliklerden kaynaklanıyor olabilir.
Genetik Miras
Gilbert sendromu farklı kalıtım kalıplarına sahip olabilir. Koşul, UGT1A1 * 28’in UGT1A1 geninin promotör bölgesinde değişmesi sonucu ortaya çıktığında, otozomal resesif bir düzende kalıtılır, yani her bir hücredeki genin her iki kopyası da mutasyona sahiptir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve semptomlarını göstermezler.
Bu durumUGT1A1 genindeki bir yanlış mutasyondan kaynaklandığı zaman , otozomal dominant bir düzende kalıtılır, bu da her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir. Crigler-Najjar sendromu olarak bilinen daha ciddi bir durum, UGT1A1 geninin her iki kopyası da mutasyonlara sahip olduğunda meydana gelir.
Kaynakça
https://ghr.nlm.nih.gov/condition/gilbert-syndrome#inheritance