Genel Bilgiler
Larsen sendromu LRS vücutta kemiklerde anormal büyümeye yol açan otozomal dominant nadir bir genetik hastalıktır. Sendrom ilk olarak 1950 yılında yayınlanan bir dergide (MD), Amerikan ortopedist Loren J. Larsen tarafından tarif edilmiştir ve ismini “Larsen” olarak almıştır. Her yıl yaklaşık 100.000 bebekten 1’ini etkileyen sendrom, aynı ailedeki çocuklarda bile, bozukluğu olan birçok farklı semptomlara neden olabilir. LarsenSendromlu kişiler normal bir zekaya sahiptir. (1) Belirtiler esas olarak eklemlerden ve iskeletlerden gelir. Bozukluğun karakteristik bulguları arasında büyük eklemlerin çıkıkları, iskelet malformasyonları, kısa boy ve ayırt edici yüz, bacak özellikleri bulunur. Sendrom kalıtsaldır ve tüm vücut dokularının gelişimi için önemli olan bir protein eksikliğinden kaynaklanmaktadır. Bu protein bağ dokusu proteini filamin B(FLNB)’dir ve sendrom buproteini kodlayan gendeki mutasyonlardan kaynaklanır. Mutasyon kendiliğinden ortaya çıkabilir veya otozomal dominant özellik olarak kalıtsal olabilir. (3) Tedavi temel olarak eklem kusurlarının düzeltilmesi ve önlenmesinden oluşur. Çoğu insan için bu sendrom, günlük yaşamda veya kariyer seçimlerinde önemli bir sınırlamaya neden olmaz. (2) Aşağıda Larsen Sendromlu bireylerin ayırt edici yüz özellikleri görülmektedir.
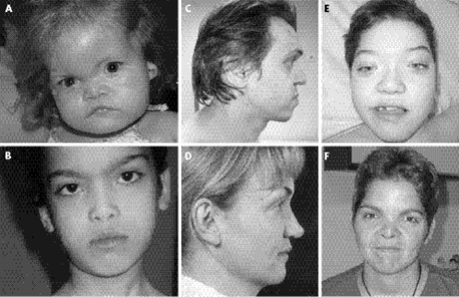
Şekil 1. Larsen Sendromu tanısı olan hastaların yüz özellikleri. (5)
Görülme Sıklığı
Larsen Sendromu, erkekleri ve kadınları eşit düzeyde etkiler. Genel popülasyondaki 100.000 kişiden 1’inde meydana geldiği tahmin edilmektedir. Larsen Sendromu tanısındaki zorluk nedeniyle, genel popülasyondaki gerçek sıklığını belirlemek zordur. (3) Orphanet epidemiyolojik verilerine göre ise bu değer Avrupa’da 1-9 / 1000000 şeklinde verilmiştir.
Belirti ve Semptomlar
Larsen Sendromlu bireylerde normalden daha geniş gözler (hipertelorizm), belirgin bir alın ve çökük köprülü burun gibi kendine özgü yüz özellikleri bulunur. Ağız çatısının (yarık damak) ya da boğazın arkasına sarkan yumuşak dokudaki yarığın (bifid uvula) tam olarak kapanmaması da etkilenen bireylerin %15’inde ortaya çıkabilir. Sağırlık genellikle kulak çınlaması (kulak çınlaması) ile yaygındır ve bireylerin %21’inde orta kulak hücrelerinin malformasyonları ile ilişkili olabilir. Klasik Larsen Sendromu olan az sayıda bireyde, trakeomalazi (soluk borusunun kıkırdakta anormal yumuşaması) bulunur. (3) Bu semptomlar aynı aile bireyleri içinde bile büyük ölçüde değişmektedir. Aşağıda Larsen Sendromu’nun genel belirti ve semptomları sıralanmıştır. (2)
• Skolyoz veya kifoz gibi omurilik deformasyonu ve servikal omurga anormallikleri
• Çıkık kalçalar, dizler ve dirsekler
• Anormal derecede gevşek eklemler
• Bilekte ve ayak bileklerinde ekstra kemikler
• Yassı, kare şeklindeki parmak uçları
• Öne çıkan bir alın, burnun yassı köprüsü, geniş gözler ve yarık damak gibi kraniyofasiyal anomaliler
• İşitme kaybı (kulaklardaki bazı kemikler düzgün şekilde oluşmamıştır)
•Solunum problemleri, kısa boy (2)
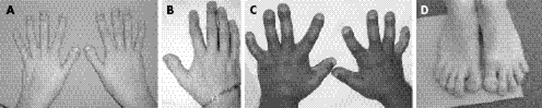
Şekil 2. Larsen Sendromlu bireylerin ayaklarının klinik görüntüleri. (5)
Kalıtım Deseni
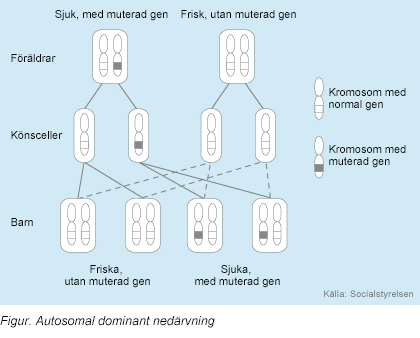
Larsen Sendromu otozomal dominant kalıtılır ve bu hastalığa kromozom 3’ün (3p14.3) kısa kolunun üzerinde bulunan FNLB adlı gendeki mutasyon sebep olur. FLNB’dekimutasyonlar, bu gen tarafından kodlanan proteinin fonksiyon bozukluğuna neden olur.Filamin B( FLNB ), aktin hücre iskeleti filamentlerini çapraz bağlayan büyük bir dimerik aktin bağlama proteinidir. Bu protein, hücrelerin şeklinin ve hacminin korunmasına yardımcı olur. Filamin B, erken fetal gelişim sırasında kas hücre liflerine bağlanarak kas oluşumunda rol oynar (embriyonik kas hücresi). Ayrıca protein filamenti A’ya (FLNA) bağlanarak, ilkel sinir hücrelerinin farklı bölgelere göç etmesini sağlar.(2)
Otozomal dominantta mutasyona uğramış genden her bir hücrede bir kopya olması belirtilerin görünmesi için yeterlidir. Bazı vakalarda hastalıktan etkilenen kişi bunu mutasyondan etkilenen ebeveyninden alır. Bazı durumlarda da mutasyon kendiliğinden ortaya çıkar.
Bazı araştırmacılar bu hastalığın otozomal resesif kalıtıldığını, etkilenmemiş ebeveyne ve etkilenmiş kardeşe sahip bireyin bu hastalıktan germline mozaizm (gonadal mozaiklik) sayesinde etkilenmediğini öne sürmektedir. Yani çok çocuklu ailelerde hasta olmayan ama yumurta ya da spermlerinde (diğer vücut hücrelerinde değil) mutasyon görünen ebeveyn hastalık yapıcı mutasyon kalıtılabilir. Bu da hastalığın otozomal resesif görünmesine neden olabilir. (3)
Otozomal resesif formda Larsen Sendromu, Afrika’nın doğu kıyılarındaki Reunion Adası’ndaki birçok büyük ailede tespit edilmiştir. Bu bozukluk da çoklu eklem çıkıklarına neden olur ancak farklı klinik ve radyolojik özelliklere sahiptir. B4GALT7’de bulunan bir mutasyondan kaynaklanmaktadır. Otozomal resesif Larsen Sendromu olan hastalarda karbonhidrat sülfotransferaz 3 (CHST3 ) genindeki mutasyonlar tanımlanmıştır. Homozigoz GZF1 varyantları şiddetli miyopi, retina dekolmanı ve tekrarlayan büyük eklem çıkıklarından etkilenen Suudi ailelerinde de bu durum bildirilmiştir. (4)
Hastalıkla İlişkili Genler
Larsen Sendromu ile ilişkili genler; FlnB (filamin B), FlnA (filamin A), CHST3 (karbonhidrat sülfotransferaz 3), GZF1 (çinko parmak proteini 1), COL7A1 (kollajen tip 7 alfa 1 zinciri), B4GALT7 (beta 1-4 galaktosiltransefarz 7) olarak gösterilmiştir. (4)
Teşhis ve Tedavi Yöntemleri
Larsen Sendromu’nun tanısı, ayrıntılı bir klinik değerlendirmeye, ayrıntılı hasta geçmişine ve karakteristik klinik ve radyolojik bulguların tanımlanmasına dayanarak yapılır. Radyografik muayene ile iskelet malformasyonları tespit edilebilir. Moleküler genetik testler FLNB gen mutasyonunun varlığını doğrulayabilir. Teşhis ile bağlantılı olarak, aileye genetik rehberlik yapılması önemlidir. Ayrıca aynı sendromlu daha fazla çocuğa sahip olma olasılığının değerlendirilmesi de gerekir. Ailedeki mutasyon biliniyorsa, birçok kalıtsal hastalık için, preimplantasyonal genetik tanılama (PGD) yapılmalıdır. Aşağıda Larsen Sendromu teşhisi için kullanılan yöntemler sıralanmıştır. (2)
• Vücuttaki organ ve yapıların ayrıntılı görüntüleri için büyük mıknatıslar, radyo frekansları ve bir bilgisayar kombinasyonu kullanan manyetik rezonans görüntüleme (MRG).
• Vücudun enine kesit görüntüleri için X ışınları ve bilgisayar teknolojisinin bir kombinasyonunu kullanan bilgisayarlı tomografi (CT) taraması.
• İki düzlemsel görüntüden 3 boyutlu modeller oluşturan görüntüleme teknolojisi EOS görüntüleme. (CT taramasından farklı olarak, çocuk dik veya ayakta dururken EOS görüntüleri alınır ve bu da ağırlık taşıyan konumlandırma nedeniyle gelişmiş tanı sağlar)
• İlaç kullanımı ve etkinliğini, biyokimyasal hastalıkları ve organ fonksiyonunu belirlemeye yardımcı olabilecek kan testleri.
Larsen Sendromu birçok vücut sistemini etkileyen bir hastalıktır. Dolayısıyla birçok tıbbi bölümün multidisipliner çalışması gereklidir. Omurga, kemik ve kasla ilgili sorunlar için ortopedist, düzenli işitme değerlendirmeleri için kulak burun boğaz uzmanı, yarık damak için plastik cerrah, herhangi bir sinir sorunu için nörolog, solunum sorunları için pulmonolog, çocuğunuzun fiziksel becerisini geliştirmek için fizyoterapistler gerekebilir. Fizik tedavi gibi cerrahi olmayan seçenekleri veya büyümeye devam ederken omurgayı sabitlemek için spinal füzyon veya büyüyen çubuklar yerleştirmek gibi cerrahi seçenekler kullanılabilir. Bunun yanında kalça ve diz protezi ihtiyaçları olabilir. (2)
Özellikle yeni doğan bebeklerde, ayakların birkaç ay sonra ameliyat edilmesi gerekir, ancak başlangıçta tedavi, ayakları kademeli olarak daha iyi bir pozisyona getirebilmek için tekrarlanan germe ve sıva işlemlerinden oluşur. Daha sonra, işlem bir ray (ortez) ile gerçekleştirilir. Bu tedaviye rağmen, ayak hatası ayarının bozulma riski veya optimum pozisyonun elde edilememesi riski vardır. Daha sonra çeşitli işlemlere ihtiyaç duyulur ve çocukluk döneminde tekrarlı cerrahi işlemlerin yapılması gerekir. Eğik sırt (skolyoz) korse ile tedavi edilebilir ancak bazen ameliyat gerekir. Eldeki hatalar el cerrahı tarafından değerlendirilir ve ele alınır ve bazen el rayları (ortezler) kullanılarak düzeltilebilir. Plastik cerrahi klinikleri ile bağlantılı olarak, dudak, çene ve damak yarıkları olan çocukların nasıl takip edileceği ve tedavi edildiği ile ilgili profesyonel ekipler vardır. Bu ekipler plastik cerrah, konuşma terapisti, çene cerrahı, diş kontrol uzmanı ve psikologu içerir. Rekonstrüktif cerrahi, burun büyümesi eksikliği ve yarık damak için uygundur ve bu hastalar da konuşma terapisi gerektirebilir. Solunum (solunum) problemleri ventilatör yardımı, özel beslenme teknikleri ve göğüs fizik tedavisi dahil destekleyici tedavi gerektirebilir. (2)
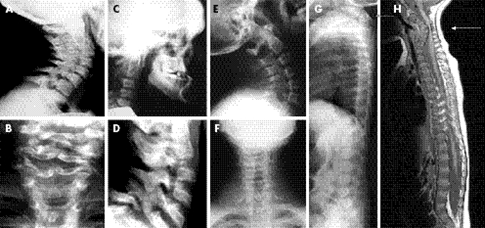
Şekil 3. Larsen Sendromlu bireylerin servikal omurga anomalileri. (5)
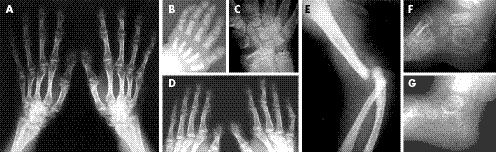
Şekil 4. Larsen Sendromlu bireylerin iskelet malformasyonlarının radyografik görüntüleri (5)
İşitme cihazını düzenli olarak kontrol etmek ve gerekirse işitme bozukluğu ve işitme cihazları için işitme uzmanıyla iletişim kurmak önemlidir. Bazı durumlarda koklear implantlar (CI) kulağın arkasındaki derinin altındaki kafatası kemiğine uygulanabilir. Koklear implant, ses sinyallerini doğrudan işitsel sinire ileten bir verici türüdür. Bu yolla, tamamen sağır olan bir çocuk konuşma ile iletişim kurmayı öğrenir. Çocuklar genellikle yaklaşık bir yaşında ameliyat edilir. Larsen Sendromlu bireylerin tedavi süreçlerinin mutlaka uzman doktorlar ve genetik danışmanlar tarafından takip edilmesi gereklidir. (2)
Kaynakça
1) Sajnani AK 1 , Yiu CK , Kral NM. (2010) “Larsen syndrome: a review of the literature and case report.” Spec Care Dentist. Nov-Dec;30(6):255-60
2) Socialstyrelsen-Göteborgs Universitete-Ovanliga Diagnoser-Larsen Syndrome
3) NORD- National Organization For Rare Disorders-Larsen Syndrome
4)Malacards-Larsen Syndrome
5) Bicknell LS 1 , Farrington-kaya Cı , Shafeghati Y , kıç P , Alanay Y , Alembik Y , Al-Madani N , Firth lH , Karimi-Nejad MH , Kim, CA , Leask K , Maisenbacher M , Moran D , Pappas JG , Prontera P , de Ravel T , Fryns JP , Sweeney E , Fritözde , Unger S , Wilson LC , Lachman RS ,Rimoin DL , Cohn DH , Krakow D , Robertson SP. (2007) “A molecular and clinical study of Larsen syndrome caused by mutations in FLNB.” J Med Genet. Feb;44(2):89-98.
6) Childrens Hospital of Phidelphia (CHOP)-Larsen Syndrome