-Nager Sendromu Çeşitleri (Nager Sendromu’nun bilinen diğer isimleri)-
- Akrofasiyal Disostoz 1 (AFD1)
- Mandibulafisayal Disostoz ‘’ TCS ‘’(Treacher Collins Tipi-uzuv anomalileriyle birlikte)
- Nager Akrofasiyal Disostoz
- NAFD Sendromu
- Preaksiyal Akrofasiyal Disostoz
- Preksiyal Mandibulofasiyal Disostoz
Genel Müzakere (Özet)
Nager sendromu,nadir görülen kalıtımsal uyuşmazlığı karakterize eden bir hastalıktır ve kronofasiyal malformasyon ile beraber başparmak, önkol anormalliklerini ortaya çıkartmaktadır .Kronofasiyal malformasyon az gelişmişlikle oluşan; elmacık kemikleri (yanak hipopalazisi) ile sonuçlanan aşağıya doğru meyilli olan palpebral fissürler oluşturmaktadır. Eksik gelişim, aşağıya sıkışmış alt çeneye (mandibular hipopalazi) neden olan anormal küçüklükte (mikroginati) ve küçük mirkotia ve/veya kusurlu (displastik) çene, yarık dudak-yarık damak adı verilen açıklıklara sebep olmaktadır (Mikroginati; solunum yolu problemlerine yol açar ve tıkanıklığa sebep olur.) Dış kulaklarda (pinrus),genellikle kulak kanalı tamamen tıkalı ya da kulak kanalı bulunmayan ve işitme bozukluğuna sebep olan anomaliler ortaya çıkartır (iletken işitme kaybı).Nager sendromu diğer sendromlardan; akrofasiyal disostoz, uzuv anomalileri, ayak başparmağı (radial kısımda) yokluğu, az gelişmiş el ya da önkol (Radius kemiği) da görülen anormal kemik füzyonları ile ayırt edilebilmektedir (radial sinostazis). Fakat el ve ayak parmakları genelde normal durumdadır.Hastaların zekaları (mental) etkilenmemiştir.Nager sendromu tipik olarak otozomal baskın gen ile (SB3F4 geni) değişiklere (mutasyonlara) neden olur.Nager sendromu birçok vakada rastgele meydana gelmesine karşın; bireyin çocuklarına yeni bir gen değişimi olarak (de novo mutasyonu) olarak iletilebilir.
Hastalığa Giriş ve Tanıtım
Nager sendromu tıp literatüründe ilk kez 1948’de Dr.Nager ve Dr.De Reynier tarafından tanımlanmıştır.Nager sendromu; akrofasiyal disostoz (AFDS) adında bilinen bir tür bozukluğa aittir.Bu bozukluklar kronofasiyal ve ekstremite anomalileri ile karakterize edilir.AFDS preaksiyal ve postaksiyal tip şeklinde iki çeşit bozulmayla ortaya çıkar.Nager sendromu preaksiyal durumda; kollarda,bacaklarda ve bu bölgelerin başparmakları üzerindeki kemiklerde (özellikle ayak başparmağı üzerinde) vücutta görülmektedir.
Hastalığın Belirti ve Bulguları
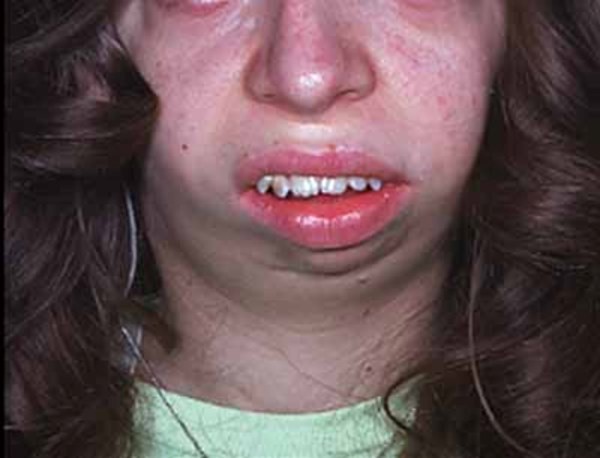
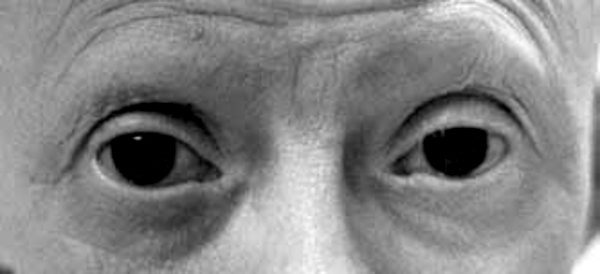
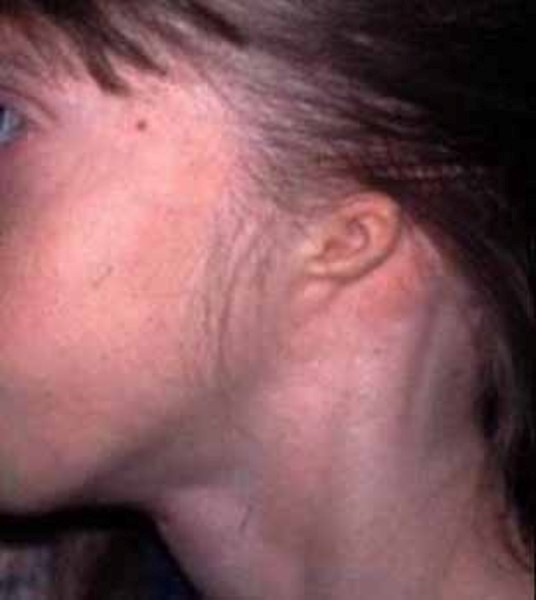
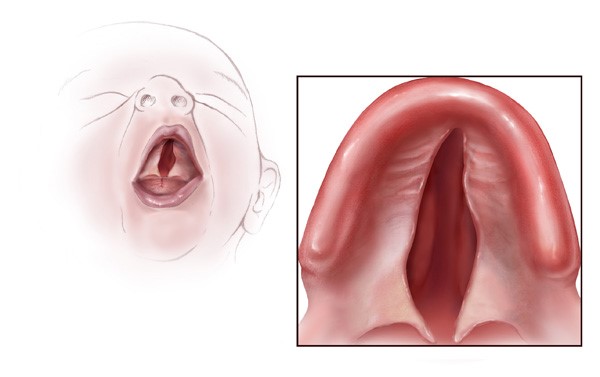
Bu tür spesifik semptomlar aynı aileden olan insanlarda meydana gelmektedir (gen aktarımı).Bu durumdan etkilenen bireylerde çeşitli kronofasiyal ve uzuv anomalileri (konjenital) genellikle fark edilmektedir. Yaygın kronofasiyal anormallikler; az gelişmiş elmacık kemiklerinde (malar hipopalazi),anormal derecede küçük bir alt çenede (mikronati),ağzın üst iç damak kısmının yeterince kapatılamamasında (yarık damak) veya velofarinjal yetmezlik (yumuşak damak,ağzın yeterince uygun şekilde kapatılamaması,konuşulamaması)nda,burun boşluğunun arkasında daralma (koanal atrezi),iç veya dış kulaklarda malformasyon,anomaliler dahilinde doku yokluğu görülmektedir.Ek olarak kronofasiyal bulgular içerisinde; gözler aşağıya sarkmaya eğimlidir (palpebral fissür), yani üst ve alt göz kapağı arasında oluşan boşluk aşağıya doğru sarkmış durumdadır.Kolobom gözler (gözlerde çatlak damar görünümü ve kızarıklık) alt göz kapaklarında kısmi veya tam olarak kirpiklerin bulunmaması ,pitozis (sarkmış göz kapakları) ve bazı hastalarda saçın elmacık kemiklerine, yanaklara kadar uzadığı görülmektedir.
Mikronati alt çene kemiğinin (mandibula) az gelişmesine neden olur. Şiddetli mandibular hipopalazi,yarık damak ve koanal atrezi ile birlikte bebeklik döneminde beslenme güçlüklerine yol açabilir.Bazı durumlarda eğer komplikasyonlar tedavi edilmez ise hayatı tehdit eden solunum sıkıntılarına sebebiyet verir.
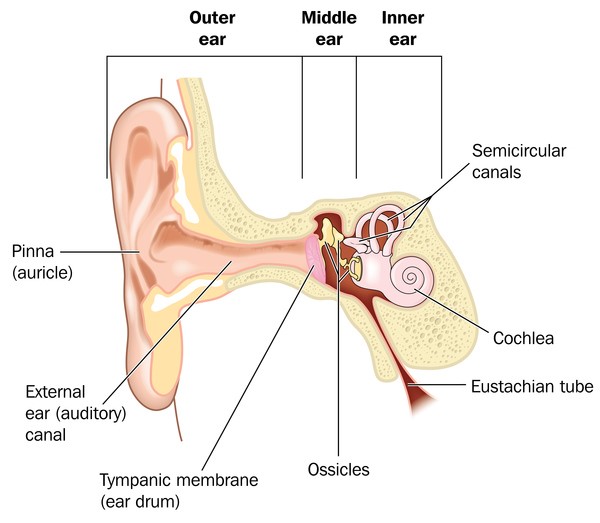
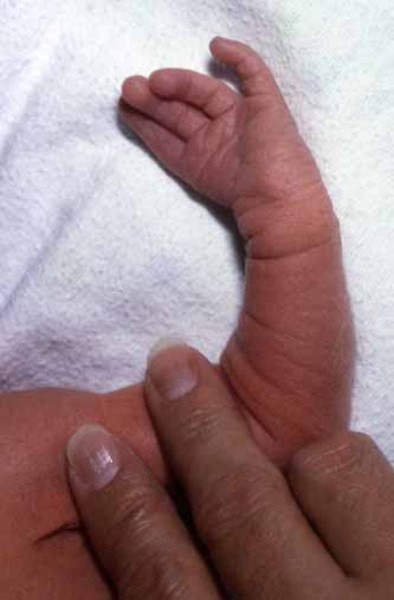
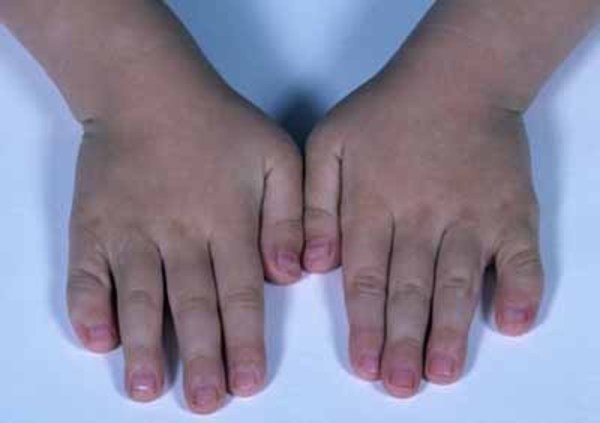
Etkilenen/hasta olan bireylerde temporomandibular eklem bozukluğu (TMJD) olabilir. Temporomandibular eklem bozukluğunda; alt ve üst çene kafanın yanında birleşir ,çenede acıya neden olur; yüz ve boyunda ağız kapatıldığında sert çene kaslarıyla birlikte üst ve alt dişlerin doğru şekilde uyuşmadığı görülür (maloklüzyon).Etkilenen bireylerde kulaklarda malformasyona (sakatlık durumu) neden olabilir ,bu durumda genelde işitme kaybı ortaya çıkmaktadır .İletken işitme kaybı;iç, orta ve dış kulakta dışardan gelen seslerin iletilmemesiyle oluşur.İşitme kaybı sorunu derecelerle çeşitlendirilebilir .İşitme bozukluğu beraberinde konuşma bozukluğunu da getirir ve geciktirir. Bireylerin Nager sendromuyla birlikte ekstra sahip oldukları anomaliler de vardır ve bu anomaliler genellikle bireyin kollarını, ellerini, ayak başparmaklarını etkilemektedir. Ayak başparmakları genellikle yoktur ya da üçüncül fazladan bir ayak başparmağı gelişmektedir. Bahsedilen başparmağın ekstra varlığında kemiğin kopyalanması (falanje) ve içinde (trifalanjel başparmak); önkol kemiğinin üstünde yan tarafta bulunan (Radius) bölgeyi etkilemektedir .Hastalarda daha az görülen anomalilerden biri de (sindaktili) yapışık parmaklılık ve parmakların (kompodaktili) bükülmüş durumda sıkışmalarıdır. Önkolun (radioulnar sinoztozis) anormal oluşumu iki ana kemikte (ulna ve Radius) yumuşak doku bağlantısı şeklinde ayrıca oluşabilir. Bu anomaliler önkolların anormal derecede kısa olmasıyla saptanabilir. Bu anomalilere sahip bireyler günlük hayatlarında kollarını tamamen düzleştiremedikleri için ve dirsek hareketleri kısıtlı olduğu için zorluk çekmektedirler. Bu anomaliden şiddetli etkilenen bireylerin ise üst uzuvları çok daha kısa durumdadır (fokomeli).Genellikle önkol ve ellerde yaygın anomaliler olduğunu bilsek de ,bazı bireyler ayak ve bacaklarında benzer duruma sahiptirler. Bu durumda daha önce de bahsedildiği gibi yapışık ayak parmakları ,ayak başparmağının içe dönmesi (hallux valgus),ayak parmaklarının bulunmaması, clubfoot görülmektedir.
Çoğu hasta Nager sendromuyla beraber sağlıklı durumdadır.Fakat sendromdan şiddetli etkilenen bireylerde ciddi içsel bozukluklar görülmektedir.Bu içsel bozukluklar özellikle böbrekleri ve/veya kalb etkilemektedir.Tıp literatüründe ek olarak nadir semptomlar raporlanmıştır.Buna örnek olarak;diafragmatik herni(diyafram fıtığı-göğüs ve karın arasında anormallik),az gelişmiş larinks (gırtlak), sebebiyle solunum problemleri,ek iskelet problemleri (ilk kaburganın az gelişmesi),omurganın anormal derecede eğriliği (skolyoz) veya kalça çıkığı gösterilebilir.
Hastalığın Nedenleri
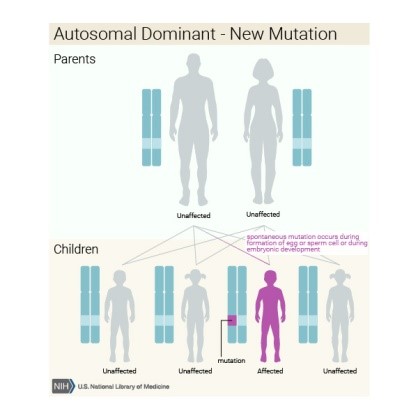
Çoğu durumlarda Nager sendromu ‘’SF3B4’’ geninde mutasyonlara sebep olur.Genler proteinleri oluşturmak için talimat ve bu durum vücudun birçok fonksiyonunda kritik bir rol oynar.Mutasyon gerçekleştiğinde genin protein ürünü hatalı,yetersiz ya da yok olmuş olabilir.Bu fonksiyonlara bağlı olarak belirtili olan protein vücuttaki birçok organ sistemini olumsuz etkileyebilir.Çünkü Nager sendromuna sahip bireyin ‘’SF3B4’’ geninin tek bir kopyasında değişiklik vardır.Bu da bizim bildiğimiz gibi Nager sendromu bireye otozomal dominant gen koşulu ile yerleşir.Çoğu durumda ise yeni bir sperm ve yumurta oluşumu sonunda ortaya mutasyon çıkar.Bu mutasyon sadece aile fertleri arasına yeni katılacak olan bireyin etkilenmesini;önceki ya da sonraki bireylerde etkilenme olmamasını sağlar.Ancak ailede Nager sendromlu ilk kişi olan birey yine de çocuklarına bu hastalığı aktarmada %50 riske sahiptir.Görünüşe göre etkilenmeyen ebeveynler için doğmuş,Nager sendromlu kardeşlerin önceki raporları, Nager sendromu için farklı bir resesif formunu temsil edebilir.Ancak orta derecede etkilenmiş bir ebeveyndeki durumu tanımadığı veya sadece yumurtalık ve testiste gen değişikliği olan bir ebeveyn olması nedeniyle daha olasıdır.(gonodal-germinal-mozaisizm)
Etkilenen Popülasyonlar
Nager sendromu kadınlarda ve erkelerde eşit olarak görülmektedir ve eşit olarak etkilemektedir.Genel popülasyonda hasalığın tam artışı ve yaygınlığı bilinmemektedir.Bu hastalık birçok vakasında yanlış teşhis edilmiş veya bir teşhis konulamamıştır;tıp literatüründe 100’den fazla vaka raporlanmış fakat genellikle hastalığın tanısının doğru frekansta belirlenmesi mümkün olamamıştır. Nager sendromu nadir bir hastalık olmasına rağmen akrofasiyal disostozdan en yaygın görülenidir.
Nager Sendromu ile İlgili Olan, Benzer Hastalıklar
Aşağıdaki yazılan hastalıkların semptomları Nager sendromunun belirtilerine benzerlik göstermektedir.Fakat ayrı bir tanı konulması için bileşimler yararlı olmaktadır.
Miller Sendromu; postaksiyal akrofasiyal disostoz olarak da bilinmektedir. Nadir bir genetik hastalıktır ve kronofasiyal malformasyonlar ile karakterize edilir .Kollarda, ellerde ve/veya ayaklarda; -genellikle postaksiyal- küçük parmak ve ayak parmağı tarafında anomaliler meydana gelmektedir .Kronofasiyal anomali; bireyde az gelişmiş elmacık kemikleri (malar hipopalazi),anormal derecede küçük bir alt çene (mikroginati),yarık dudak,küçük ve çıkıntılı kepçe kulalar ve/veya doku eksikliği, kolobomlara (düşük göz kapakları) sebebiyet vermektedir .Uzuvlarda anomalilere; eksik gelişme, ayak parmaklarında tek düzelik ve/veya yanlış gelişme ,kollarda anormal kemik füzyon oluşumu (radioulnar sinoztozis), kollarda alışılmadık derecede kısa görünüm oluşturmaktadır. Bazı durumlarda ek olarak fiziksel anomaliler ortaya çıkmaktadır fakat zeka bu durumdan etkilenmez. Miller sendromu Nager sendromuna bu semptomlarıyla çok benzese de asıl farklılık farklı genlerde mutasyonlar olması sonucuyla belirlenir. Miller sendromu; mutasyonların ‘’DHODH’’ geninde neden olduğu özellikler sayesinde otozomal resesif olarak miras alınır.(genler sayesinde aktarılır).
Treacher Collins Sendromu; nadir bir genetik hastalıktır ve kafa, yüz anomalileriyle Nager sendromundan (hipopalazik yüz yapıları, çene ve elmacık kemiği yakınındaki yapılar da dahil olmak üzere –elmacık kemiği kompleksi- ) sayesinde ayırt edilmektedir. Ayrıca bireyin uzuvları da normal durumdadır. Kronofasiyal anomalilerde ağızda, gözlerde veya çenede; elmacık kemiklerine dahil olma eğilimi bulunmaktadır.Ek olarak çeşitli yüz anomalileri; hastalıktan etkilenen bireyde kusurlu dış kulaklar ve orta kulak, göz (oküler) anomalileri; aşağıya doğru sarkmaya meyilli göz kapakları ve palpebral fissür meydana gelmektedir. Ayrıca hastalıktan etkilenen bireylerde işitme kaybı ve nefes alma (solunum) zorlukları gelişmektedir. Zeka ve davranışsal anomalilerde; raporlanmış bir durumun parçası olarak mikrosefali ve psikomotor gecikmeleri (delay) görülmektedir. ‘’TCS’’ ile ilişkili spesifik semptomlar ve fiziksel özellikler bir kişiden diğerine çok büyük ölçüde olabilir. Hastalığın tanısı konulmadan ve semptomları hafifken diğer belirtiler çok ciddi gelişebilir; hayatı tehdit eden ciddi bir solunum komplikasyonları ortaya çıkabilir. TCS; ‘’TCOF1, POLR1C, POLR1D’’ genlerinde meydana gelen bir mutasyondan kaynaklanır. TCOF1 veya POLR1D durumunda kalıtım şekli otozomal dominantken,POLR1C durumunda kalıtım otozomal resesiftir.
Rodriguez Sendromu; Rodriguez sendromu, Rodriguez’in akrofasiyal disostozu olarak da bilinir ve son derece nadir görülen bir hastalıktır. Rodriguez ve Nager sendromlarında da uzuv ve kronofasiyal anomalileri olarak ciddi bir benzerlik bulunmaktadır. Bazı araştırmacılar Rodriguez Sendromuna ait vakaların, Nager sendromunun daha ciddi bir ifadesi olabileceğine inanmaktadır. Ek olarak fiziksel bulgular çoğunlukla Nager sendromu ile birlikte bulunur. Rodriguez sendromlu bireylerde; omuz ve pelvik eklem desteği az gelişmiş durumdadır; kalp, merkezi sinir sistemi ve ürogenital sistemlerinde anormallikler meydana gelmiştir. Rodriguez sendromlu bebekler genellikle ölü doğarlar ya da yenidoğan dönemlerinde şiddetli solunum yolu problemleri nedeniyle direkt ölmektedirler. Rodriguez sendromunun kalıtımsal olduğu düşünülüyordu fakat son zamanlarda bir çocukta Rodriguez sendromuyla birlikte, SB3F4 geninin bir kopyasının de novo mutasyonuna uğradığı görüldü ( Nager sendromunda olduğu gibi). Çeşitli nadir bozukluklar; Wayers akrofasiyal disostoz, Catania akrofasiyal disostoz ve Palagnia akrofasiyal disostoz postaksiyal ve akrofasiyal disostoz bulgularını içermektedir. Diğer bozukluklar çok fazla semptomlara sahip POLR1C durumunda r ya da Nager sendromunun fiziksel bulgularıyla örtüşmektedir. Bunlara örnek; Ophthalmo akromelik sendromu, Pallister Hall sendromu, Mandibulafasiyal disostoz tipi Guion-Almeida, Burn-Mckeownsendromu ve Oculo-Auriculo-Vertebral sendromu gösterilebilir.
Teşhis
Nager sendromunun tanısı uzun süre düşünülmüş bir klinik evrime dayanır, detaylı hasta öyküsü ve karakteristik fiziksel bulguların belirlenmesiyle açıklanabilir. Bu hastalıkla ilişkili anormalliklerin çoğu doğumda bulunur (konjenital). Moleküler genetik testleri Nager sendromunun tanısının doğru yapılmasını sağlar. Moleküler genetik testleri SF3B4 genindeki mutasyonu tespit edebilir ama bu uygulama sadece uzman laboratuvarlarda gerçekleşmektedir.
Klinik Test ve Çalışma
Özel röntgen çalışmaları hastalığın varlığını onaylar ve/veya kesin olarak kronofasiyal anormallikler tarafından belirlenir.Örneğin bu tür görüntüleme testleri; anormal derecede küçük olan çeneyi (mikroginati) , gelişmemiş olması sebebiyle alt çene kemiğini (mandibular hipopalazi) ve az gelişmiş elmacık kemiğini (malar hipopalazi) göstermektedir.
Standart Uygulanan Terapiler ve Hastalığın Tedavisi
Nager sendromunun tedavisi her bireyde görülen belirgin spesifik semptomlara göre uygulanır.Tedavi uzman ekibin koordineli çalışmasını gerektirir.Çocuk doktorları, ağız cerrahisinde uzmanlar, plastik cerrahi uzmanları, pediatrik kulak-burun-boğaz uzmanları (pediatrik KBB), göz bozukluklarının teşhis ve tedavisinin uzmanları (göz doktorları) kulak bozukluklarının teşhis (KBB Hekimleri), işitme kaybının tedavisinde uzmanlar (odyologlar), psikologlar ve diğer sağlık hizmeti profesyonelleri ; hasta bireyin tedavisini sistematik ve kapsamlı bir şekilde planlamalıdır.Hastalıktan etkilenen bireyler kraniyofasiyal merkeze yönlendirilmekten yararlanabilirler. Özel tedavi olarak; boğazda küçük bir kesik ile açıklık oluşturduktan sonra, boğaza belirli bir boyutta kesiğe uygun tüp yerleştirilir v, buradaki amaç hastanın solunum yolu açıklığını sağlamaktır (trakeostomi). Bireyin doğru beslenmesini sürdürebilmek için de cerrahi girişim gerekli olabilir ; midede küçük bir açıklık oluşturularak beslenme tüpü içeriye doğru salınır. Çenedeki, gözlerdeki ve uzuvlardaki anomalileri düzeltebilmek için cerrahi girişim sağlanmalıdır. Yarık damak ve yarık dudak mevcutsa eğer bu durumda ameliyat ve konuşma terapisi yapılır. Kaburgalarda oluşan anomalilergibi iskelet malformasyonlarında , dirsek hareketinin ve kolların hareketinin kısıtlılığında, skolyozda cerrahi işlem uygulanmalıdır. Konjenital (doğumsal) kalp sorunlarında/hastalıklarında da ameliyat durumu gereklidir. Erken müdahale ile uygun fiziksel,mesleki ve konuşma terapisi servisleri çocuklarda tam potansiyele ulaşmayı sağlatıyor.Fiziksel terapiler, yürüyüşlerinde yardımcı olmak için gerekli olabilir.Konuşma terapisi ise; işitme kaybı sebebiyle konuşma gelişimi gecikmesi olan bireylerin konuşmalarını sağlar.İşitme kaybı kulaklara tüplerin takılmasını gerektirebilir veya bir işitme cihazı kullanılmasını gerektirebilir. Etkilenen bireylere genetik danışmanlık tavsiye edilmektedir, aileleri de bu öneriden yararlanmalıdır. Tüm aile içinse aslında en gerekli olan psikososyal bir destektir.
Kaynaklar
2-)https://rarediseases.info.nih.gov/diseases
3-)https://www.orpha.net./consor/cgi-bin/Disease_Search.php
4-)https://ghr.nlm.nih.gov/condition
5-)https://rarediseases.org/for-patients-and-families/information-resources/rare-disease-information/