Genel Bilgi
Trisomy 13, normal 2 kopya yerine, vücut hücrelerinde 3 kopya kromozom 13’ün bulunmasıyla karakterize edilen bir tür kromozom bozukluğudur. Etkilenen bazı insanlarda, hücrelerin sadece bir kısmı ekstra kromozom 13’ü (mozaik trizomi 13 olarak adlandırılır) içerirken, diğer hücreler normal kromozom çiftini içerir.Trisomy 13 ciddi zihinsel sakatlığa ve doğuştan kalp defekti gibi birçok fiziksel anormalliklere neden olur; beyin veya omurilik anormallikleri; çok küçük veya zayıf gelişmiş gözler (mikroftalmi); ekstra parmak veya ayak parmakları; yarık damak veya damaksız yarık dudak; ve zayıf kas tonusu (hipotoni). Vakaların çoğu kalıtsal değildir ve sağlıklı ebeveynlerde yumurta veya sperm oluşumu sırasında rastgele bir hatadan kaynaklanmaktadır. Hayatı tehdit edici çeşitli tıbbi problemler nedeniyle, trizomi 13’ü olan birçok bebek, yaşamın ilk günleri veya haftaları boyunca hayatta kalamaz.
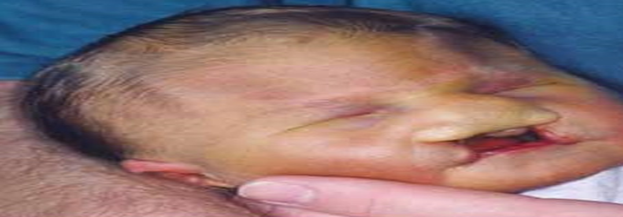
Şekil.1. Patau Hastası Bir Birey (Yarık Dudak)
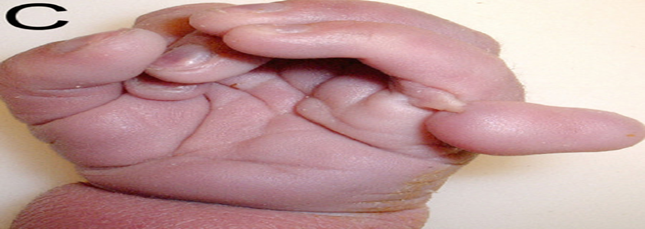
Şekil.2. Patau Hastası Bir Bireyin Eli (Ekstra Parmak)
Genetik Değişiklikler ve Etken Faktörler
Trisomy 13 Sendromu olan bireylerde, tümü veya nispeten büyük bir kromozom 13 bölgesi, hücrelerde iki kez değil üç kez (trisomi) bulunur. Vakaların yaklaşık yüzde beşinde, sadece yüzde bir hücre, ekstra 13. kromozomu (mozaikçilik) içerir. Kromozomlar tüm vücut hücrelerinin çekirdeğinde bulunur. Her bireyin genetik özelliklerini taşırlar. İnsan kromozom çiftleri, erkekler için eşit olmayan bir X ve Y kromozomu çifti ve kadınlar için iki X kromozomu ile 1 ila 22 arasında numaralandırılmıştır. Her kromozomun “p” olarak adlandırılan kısa bir kolu ve “q” harfi ile tanımlanan uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış bantlara bölünmüştür. Belirli bir bölge veya kromozom 13 bölgelerinin tizomisi (veya “çoğaltma”), bozukluğu karakterize eden semptom ve bulgulardan sorumludur. Semptomların şiddeti ve aralığı, kromozomun kopya kısmının uzunluğuna ve konumuna bağlı olabilir. Ek olarak, trizomi 13 mozaikçiliği olanlar tipik olarak daha az ciddi semptomlara sahiptir; Bununla birlikte, bu gibi durumlarda, hastalık belirtileri normalden neredeyse tüm malformasyon spektrumuna kadar değişen değişkenlik gösterebilir. Trisomy 13 Sendromu olan çoğu kişide, kromozom 13’ün çoğalması, ebeveynlerden birinin (örneğin mayoz bölünmesi sırasında yoksun olma) üreme hücrelerinin bölünmesi sırasındaki spontan (de novo) hatalarından kaynaklanır. Kanıtlar, ileri yaşta ebeveyn yaşı ile birlikte bu tür hataların riskinin artabileceğini göstermektedir. Hücrelerin sadece bir yüzdesinin trizomi 13 anormallik (mozaiklik) içerdiği durumlarda, döllenmeden sonra hücre bölünmesi sırasında da hatalar oluşabilir (mitoz). Etkilenen bireylerin yaklaşık yüzde 20’sinde, trizomi 13, kromozom 13 ve başka bir kromozom içeren bir translokasyondan kaynaklanır. Translokasyonlar, belirli kromozom bölgeleri kopup yeniden düzenlendiğinde meydana gelir, bu da genetik materyalin ve değiştirilmiş bir kromozom setinin kaymasına neden olur.
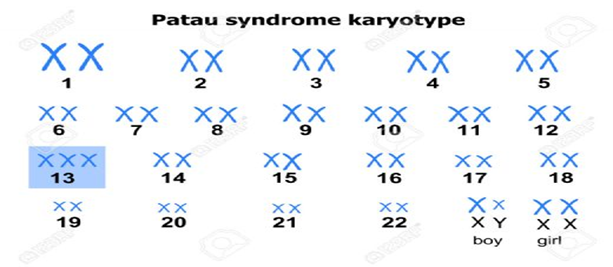
Şekil.3. Patau Hastası Bir Bireyin Karyotip Analizi
Belirti ve Semptomlar
Trisomy 13 Sendromu olan bireylerde, ilişkili semptomların ve bulguların kapsamı ve ciddiyeti, kromozom 13’ün kopyalanmış (trisomik) kısmının spesifik konumuna ve ayrıca anormalliği içeren hücrelerin yüzdesine bağlı olabilir. Bununla birlikte, etkilenen birçok bebek ve çocukta, bu anormallikler gelişimsel gecikmeler, derin zihinsel gerilik, olağandışı küçük gözler (mikroftalmi), üst dudaktaki anormal bir oluk (yarık dudak), ağzın çatısının kapanmaması (yarık damak) olabilir. ), etkilenen erkeklerde inmemiş testisler (kriptorşidizm) ve ekstra (süpernumerary) parmak ve ayak parmakları (polidaktili). Baş ve yüz (kraniyofasiyal) alanın ek malformasyonları da mevcut olabilir, örneğin eğimli bir alına sahip olan nispeten küçük bir kafa (mikrosefali); geniş, düz bir burun; yaygın olarak belirlenmiş gözler (oküler hipertelorizm); gözleri örten dikey cilt kıvrımları; iç köşeler (epicanthal kıvrımları); kafa derisi kusurları; ve hatalı biçimlendirilmiş, alçak ayarlanmış kulaklar. Etkilenen bebekler ayrıca beynin belirli bölgelerinin (örneğin, ön beyin) eksik gelişimini de sağlayabilir; böbrek (böbrek) malformasyonları; ve doğumda yapısal kalp (kalp) kusurları (doğuştan). Örneğin, karakteristik kalp defektleri, kalbin üst veya alt odalarını (atriyal veya ventriküler septal defektler) bölen bölümdeki anormal bir açıklığı veya iki ana arter (aort, pulmoner arter) arasında ortaya çıkan iki ana arter arasındaki fetal açıklığın sürekliliğini içerebilir kalp (patent duktus arteriosus). Trisomy 13 Sendromlu birçok bebek beklenen oranda büyümez ve kilo alamaz (gelişemez) ve şiddetli beslenme güçlüğü, azalan kas tonusu (hipotoni) ve spontan beratlamanın (apnenin) geçici olarak kesildiği bölümler vardır. Bebeklik döneminde veya erken çocukluk döneminde hayatı tehdit eden komplikasyonlar gelişebilir.
Genetik Görülme Sıklığı
Trisomy 13 Sendromu bazen sendromun 1960 yılında trizomik kökenini tanımlayan araştırmacılardan (Patau K) sonra, Patau Sendromu olarak adlandırılır. Sendrom, kadınları erkeklerden biraz daha sık etkiler ve yaklaşık 5.000 ila 12.000 canlı doğumda görülür. Kanıtlar, tanınan tüm düşüklerin yaklaşık yüzde birinin, Trisomy 13 Sendromu ile birlikte gerçekleştiğini göstermektedir. Ek olarak, yukarıda belirtildiği gibi, Trisomy 13’ün sıklığı annenin ilerleyen yaşı ile birlikte artar.
Kalıtım Paterni/Deseni
Çoğu trizomi 13 vakası kalıtsal
değildir ve sağlıklı ebeveynlerde yumurta ve sperm oluşumu sırasında rastgele
olaylardan kaynaklanır. Hücre bölünmesinde bağlanma denilen bir hata, anormal
sayıda kromozom içeren üreme hücresi ile sonuçlanır. Örneğin, bir yumurta veya
sperm hücresi, ekstra bir kromozom 13 kopyası alabilir. Bu atipik üreme
hücrelerinin biri, bir çocuğun genetik yapısına katkıda bulunursa, çocuğun
vücudun her hücresinde ekstra bir kromozom 13 olacak.
Translokasyon trizomi 13 miras edilebilir. Etkilenmemiş bir kişi, 13 nolu
kromozom ile bir başka kromozom arasında genetik materyalin yeniden
düzenlenmesini taşıyabilir. Bu yeniden düzenlemelere dengeli translokasyonlar
denir, çünkü kromozom 13’ten fazla bir materyal yoktur. Kromozom 13 içeren
dengeli bir translokasyona sahip bir kişi, kromozom 13’ten çocuklarına ekstra
materyal geçirme şansını arttırır.
Teşhis Yöntemi ve Tedavi
Bazı durumlarda, doğumdan önce (doğum öncesi) fetal ultrasonografi, amniyosentez ve / veya koryon villus örneklemesi (CVS) gibi özel testlerle Trisomy 13 Sendromu tanısı önerilebilir. Fetal ultrasonografi sırasında, yansıyan ses dalgaları gelişen bir fetüsün görüntüsünü oluşturarak, potansiyel olarak bir kromozomal bozukluk veya başka anormallikler önerebilecek bulguları ortaya çıkarır. Trisomy 13 Sendromu tanısı doğumdan sonra (doğum sonrası) ayrıntılı bir klinik değerlendirme, karakteristik fiziksel bulguların tespiti ve kromozomal analiz ile konulabilir. Testler ayrıca, embriyonik ve / veya fetal hemoglobinin Trisomy 13 Sendromlu yenidoğan ve bebeklerin kanında olağandışı kalıcılığını ortaya çıkarabilir. (Hemoglobin, kırmızı kan hücrelerinin oksijen taşıyan bileşenidir.) Sendromu olan bebekler için, Trisomy 13 Sendromu ile potansiyel olarak ilişkili koşulların erken tespitini ve uygun yönetimini sağlamak için dikkatli izleme ve çeşitli özel testler yapılabilir. Trisomy 13 Sendromu’nun tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Böyle bir tedavi multidisipliner bir tıp uzmanları ekibinin koordine çabalarını gerektirebilir. Bazı durumlarda, önerilen tedavi, hastalıkla ilişkili bazı anormalliklerin cerrahi olarak düzeltilmesini içerebilir. Yapılan cerrahi işlemler anatomik anormalliklerin doğasına ve ciddiyetine, bunlarla ilişkili semptomlara ve diğer faktörlere bağlı olacaktır. Bu bozukluğu olan çocuklar için destek ekibi yaklaşımı faydalı olabilir ve fizik tedavi, tıbbi ve / veya sosyal hizmetleri içerebilir. Genetik danışmanlık, Trisomy 13 Sendromu olan çocukların aileleri için de faydalı olacaktır. Bu hastalığın diğer tedavisi semptomatik ve destekleyicidir.
Hastalığın Diğer İsimler
- Bartholin-Patau sendromu
- D Trisomy Sendromu
- Kromozom 13
- Trisomy 13
Kaynakça
[1] https://rarediseases.info.nih.gov/diseases/7341/trisomy-13
[2] https://rarediseases.info.nih.gov/diseases/7341/trisomy-13
[3] https://rarediseases.org/rare-diseases/trisomy-13-syndrome/
Görsel Kaynakça