Genel Bilgi
Polikistik böbrek hastalığı (PKD) , böbrekleri ve diğer organları etkileyen bir hastalıktır. Kist adı verilen sıvı dolu keseler kümeleri böbreklerde gelişir ve bu kistler böbreklerin kandan atık ürünleri filtreleme yeteneklerine müdahale eder. Kistlerin büyümesi böbreklerin genişlemesine neden olur ve böbrek yetmezliğine yol açabilir. Kistler ayrıca diğer organlarda, özellikle de karaciğerde gelişebilir.
Polikistik böbrek hastalığının sık görülen komplikasyonları tehlikeli derecede yüksek kan basıncı ( hipertansiyon ), sırt veya yanlarda ağrı, idrarda kan (hematüri), tekrarlayan idrar yolu enfeksiyonları, böbrek taşları ve kalp kapağı anormalliklerini içerir. Ek olarak, polikistik böbrek hastalığı olan kişilerde anormal şişkinlik ( anevrizma) riski daha yüksektir (aort denilen büyük bir kan damarı içinde veya beynin tabanındaki kan damarlarında). Anevrizmalar yırtılırsa hayatı tehdit edici olabilir.
Polikistik böbrek hastalığının iki ana formu , normal başlangıç yaşı ve aileden geçme şekli ile ayırt edilir. Otozomal dominant form (bazen ADPKD olarak adlandırılır) tipik olarak yetişkinlikte başlayan belirti ve semptomlara sahiptir, ancak böbreklerdeki kistler sıklıkla doğumdan veya çocukluktan itibaren mevcuttur. Otozomal dominant polikistik böbrek hastalığı , genetik nedene bağlı olarak tip 1 ve tip 2’ye ayrılabilir. Polikistik böbrek hastalığının otozomal resesif formu (bazen ARPKD olarak adlandırılır) daha nadirdir ve genellikle yaşamın erken dönemlerinde ölümcüldür. Bu durumun belirti ve semptomları genellikle doğumda veya erken bebeklik döneminde belirgindir.
Otozomal (veya İnfantil) resesif polikistik böbrek hastalığı (ARPKD), böbreklerde sıvı dolu keseler (kistler) oluşumu ile karakterize nadir bir genetik hastalıktır. Etkilenen bebeklerin çoğu yenidoğan (yenidoğan) döneminde böbrekleri büyütmüştür ve bu sırada bazı vakalar ölümcül olabilir. ARPKD basit bir böbrek hastalığı değildir ve vücudun ek organ sistemleri de özellikle karaciğerde etkilenebilir. Yüksek tansiyon (hipertansiyon), aşırı susuzluk, sık idrara çıkma ve beslenme güçlüğü de oluşabilir. Etkilenen bazı çocuklar da belirgin yüz özelliklerine sahip olabilir ve solunum yetersizliğine neden olan akciğerlerde (pulmoner hipoplazi) tam gelişmemiş olabilir. Hastalığın ciddiyeti ve ortaya çıkan spesifik semptomlar bir kişiden diğerine büyük ölçüde değişebilir. Etkilenen bazı çocuklar, yaşamın ilk on yılında bir süre sonra son dönem böbrek yetmezliği geliştirir. Bazı hastalarda, ergenliğe veya hatta yetişkinliğe kadar semptomlar gelişmez.
Şu anda, PKD için bir tedavi yoktur. Ancak, semptomları kontrol altına almak, kistlerin büyümesini yavaşlatmaya ve PKD’li insanlarda böbrek fonksiyon kaybını önlemeye veya yavaşlatmaya yardımcı olmak için birçok destekleyici tedavi uygulanabilir.
Hastalığı çocuklarına geçirmekten endişe duyan PKD’li bireyler, aile planlaması konusunda kendilerine yardımcı olmak için bir genetik danışmanına danışmak isteyebilirler.
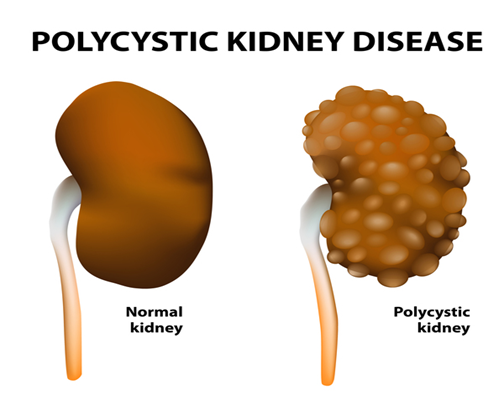
Görsel Kaynak: https://ghr.nlm.nih.gov/art/large/normal-and-polycystic-kidneys.jpeg
{kind=link}
Genetik Değişiklikler / Etken Faktörler
Çoğu polikistik böbrek hastalığı vakasında otozomal dominant kalıtım paterni vardır. Bu durumu olan insanlar , her hücrede bir PKD1 veya PKD2 geninin mutasyona uğramış bir kopyasıyla doğarlar . Bu vakaların yaklaşık yüzde 90’ında, etkilenen bir kişi etkilenen bir ebeveynin mutasyonunu devralır. Vakaların diğer yüzde 10’u yeni bir mutasyondan kaynaklanıyor Genlerin birinde ve ailesinde düzensizlik öyküsü olmayan kişilerde de ortaya çıkar.
Her hücrede bir genin değiştirilmiş bir kopyası bozukluğa neden olmak için yeterli olsa da , PKD1veya PKD2 geninin ikinci kopyasındaki ek bir mutasyon kistlerin daha hızlı büyümesini sağlayabilir ve hastalığın şiddetini artırabilir. Kistlerin genişleme ve böbrek fonksiyon kaybına neden olma hızı geniş ölçüde değişir ve tanımlanmamış diğer genlerdeki mutasyonlardan etkilenebilir.
Polikistik böbrek hastalığı da otozomal resesif paternde kalıtsal olabilir. Durumun bu formu olan insanlar , her hücrede PKHD1 geninin iki değiştirilmiş kopyası vardır . Otozomal resesif bozukluğu olan bir çocuğun ebeveynleri etkilenmez fakat değiştirilmiş genin bir kopyasının taşıyıcısıdır.
PKD1 , PKD2 , ve PKHD1 genleri mutasyonları polikistik böbrek hastalığına neden olur. PKD1 veya PKD2 genindeki mutasyonlar, otozomal dominant polikistik böbrek hastalığına neden olabilir ; PKD1 gen mutasyonları, ADPKD tip 1’e ve PKD2 gen mutasyonları , ADPKD tip 2’ye neden olmaktadır. Bu genler, fonksiyonları tam olarak anlaşılmayan proteinlerin yapılması için talimatlar sağlar. Araştırmacılar, kimyasal sinyalleri hücrenin dışından hücrenin çekirdeğine iletmekte yer aldıklarına inanıyor. İki protein normal böbrek gelişimi, organizasyonu ve işlevini desteklemek için birlikte çalışır. PKD1 veya PKD2 genindeki mutasyonlar binlerce kistin oluşumuna yol açar, böbrek ve diğer organların normal fonksiyonlarını bozan. PKD2 geninde mutasyon olan insanlar , özellikle kadınlar, tipik olarak, PKD1 mutasyonları olan insanlardan daha az şiddetli bir hastalık formuna sahiptir . Böbrek fonksiyonlarında bir düşüş de dahil olmak üzere belirti ve semptomlar, yetişkinlerde daha sonra PKD2 mutasyonu olan kişilerde ortaya çıkma eğilimindedir .
Mutasyonlar PKHD1 gen neden otozomal resesif polikistik böbrek hastalığı . Bu gen, tam işlevi bilinmeyen bir proteini yapmak için talimatlar sağlar; Bununla birlikte, protein muhtemelen hücrenin dışından hücre çekirdeğine kimyasal sinyaller iletir. Araştırmacılar, PKHD1 genindeki mutasyonların polikistik böbrek hastalığının karakteristik sayısız kist oluşumuna neden olduğunu belirlemedi .
Polikistik böbrek hastalığı genellikle genetik bir hastalık olmasına rağmen , vakaların küçük bir yüzdesi gen mutasyonlarından kaynaklanmamaktadır. Bu vakalara kazanılmış polikistik böbrek hastalığı denir . Hastalığın bu şekli en çok birkaç yıl boyunca hemodiyalizle tedavi edilen diğer böbrek hastalığı tipleri olan insanlarda görülür (kandaki atık ürünleri filtreleyen bir prosedür).
Belirti ve Semptomlar
ARPKD’nin şiddeti ve ilerlemesi, aynı ailenin üyeleri arasında bile bir kişiden diğerine büyük farklılıklar gösterebilir. Ağır vakalarda, ARPKD bebeklik döneminde hayatı tehdit eden komplikasyonlara neden olabilir. Diğer durumlarda, etkilenen bireyler, çocukluk veya ergenlik döneminde daha sonraya kadar semptom geliştirmeyebilir.
Bazı çocuklar çocukluk döneminin başlarında böbrek nakli yapılmasına ihtiyaç duyabilir; Diğerleri erken yetişkinliğe kadar nakil gerekmeyebilir veya hiç olmayabilir. Nadir durumlarda, bireyler genç erişkinliğe kadar semptom geliştirmeyebilir.
Genellikle, ilk başta hiçbir belirti yoktur. Daha sonra belirtiler arasında;
- Sırt ve alt taraflarda ağrı
- Baş ağrısı
- İdrarda kan
Genellikle, yaşamın ilerleyen dönemlerinde ARPKD gelişen kişilerde daha hafif böbrek hastalıkları, fakat daha şiddetli karaciğer hastalığı olur.
ARPKD hakkındaki tıbbi literatürün çoğu, özellikle ARPKD hastalık geninin tanımlanmasından önce yazılanlar, orantısız bir şekilde en ağır vakalara odaklanmıştır. Bu nedenle, literatürün çoğu ARPKD’nin üniform olarak ölümcül veya zayıflatıcı bir hastalık olduğu izlenimini verebilir. Araştırmacılar artık ARPKD vakalarının hafif ila şiddetli olabileceğini biliyorlar. Sonuç olarak, etkilenen bireylerin aşağıda belirtilen tüm belirtilere sahip olmayacağını not etmek önemlidir. Etkilenen bireyler kendi özel durumları, ilişkili semptomları ve genel prognozları hakkında doktorları ve sağlık ekipleriyle konuşmalıdır.
- ARPKD’nin karakteristik bulgusu böbreklerde sıvı dolu keseler (kistler) gelişmesidir. Etkilenen tüm bireyler böbreklerde kist gelişir, ancak kist gelişiminin sayısı, büyüklüğü, ilerlemesi ve ciddiyeti bir kişiden diğerine büyük farklılıklar gösterir. Etkilenen bireylerin çoğunda, böbrek kistleri uteroda büyür ve çoğalır, böbreklerde anormal genişlemeye neden olur. Büyümüş böbrekler doğumda veya yenidoğan döneminde belirgin olabilir. Bu bebeklerde böbrekler sert ve her iki yanda da hissedilir (elle hissedilebilir). Kistik böbreklerle ilişkili ek semptomlar arasında yüksek tansiyon (hipertansiyon) ve yan ağrısı vardır. ARPKD’li çocuklarda yüksek tansiyon yaygındır ve yaygın, şiddetli ve yönetimi zor olabilir.
- Ağır ARPKD vakalarında, etkilenen bebekler doğumdan kısa bir süre sonra, özellikle solunum (solunum) yetersizliği veya yetersizliği ile hayatı tehdit eden komplikasyonlarla karşılaşabilir. Solunum güçlüğü genellikle hamilelik sırasındaki yetersiz amniyotik sıvı seviyesinden (oligohidramnios) kaynaklanmaktadır. Akciğerlerin doğru gelişimini önleyen kitlesel genişlemiş böbrekler, solunum yetmezliği veya yetersizliğine de katkıda bulunabilir. Her ne kadar bazı çocuklar yenidoğan döneminde hayatta kalmasalar da (tahminlerin çoğuna göre yaklaşık yüzde 30), çocukların çoğunluğu yaşayabilir.
- Yenidoğan döneminin ötesinde hayatta kalan çocuklar, böbreklerin boyutlarını küçültebilse de, genellikle kötüleşen böbrek fonksiyonlarını geliştirirler. Çocukların çoğu, geç çocukluk, ergenlik veya genç erişkinliğe kadar kronik böbrek yetmezliği geliştirmez. Böbrek yetmezliği, böbreklerin temel işlevlerini yerine getirme yeteneğinin azalması anlamına gelir. Böbrekler, göğüs kafesinin altına yerleştirilmiş iki fasulye şeklinde organdır. Böbrekler, atık ürünlerin kandan ve vücuttan süzülmesi ve atılması, belirli hormonların yaratılması ve vücuttaki potasyum, sodyum, klorür, kalsiyum ve diğer elektrolitler gibi bazı kimyasalların dengesinin korunmasına yardımcı olmak gibi çeşitli fonksiyonlara sahiptir. ARPKD’de böbreklere verilen hasar yavaşça ilerleyici olabilir ve halsizlik, iştahta değişiklikler, şişlik, sırt ağrısı gibi çeşitli semptomlara neden olabilir, zayıf sindirim, aşırı susama ve sık idrara çıkma. Sonunda, birçok çocuk böbrek hastalığına son aşamada ilerler.
- Şiddetli ARPKD vakalarında, etkilenen bebeklerde aşırı derecede büyümüş böbrek vardır ve doğumda idrar üretimini azaltır. Uterodaki azalmış idrar üretimi, gelişen bir fetüsü çevreleyen sıvı olan amniyon sıvısının (oligohidramnios) eksikliğine katkıda bulunur. Bir fetusun korunmasına ve desteklenmesine ek olarak, amniyotik sıvı büyüme faktörlerini ve fetal gelişim için hayati olan diğer maddeleri içerir. Düşük amniyotik sıvı seviyeleri akciğer gelişimini bozabilir ve sonuç olarak, etkilenen bazı bebeklerde az gelişmiş akciğerler (pulmoner hipoplazi) olabilir. Bu bebekler yenidoğan döneminde ciddi, hayatı tehdit edici solunum (solunum) komplikasyonları yaşayabilir. Oligohidramniyoslu bebekler ayrıca derin gözler, düz bir burun, küçük bir çene (mikrognati) ve anormal, düşük ayarlı kulaklar gibi belirgin yüz özellikleri de geliştirebilir.
- Böbreklere ek olarak karaciğer, ARPKD’li çocuklarda ve yetişkinlerde de yaygın olarak etkilenir. Karaciğer vücutta gıdaları enerjiye ve besin maddelerine dönüştürmek, vitamin depolamak ve vücuttaki toksinleri süzmek dahil olmak üzere birçok işlevi yerine getirir. ARPKD’li çocuklar, aşırı lif benzeri bağ dokusunun karaciğere yayıldığı konjenital hepatik fibrozis olarak bilinen bir karaciğer hastalığı geliştirir. Her ne kadar çocuklarda konjenital hepatik fibrozis olsa da, tüm çocuklar karaciğer fonksiyon bozukluğu geliştirmez. ARPKD’de ortaya çıkabilecek karaciğer anomalileri, karaciğerin genişlemesini (hepatomegali), karaciğerden safra taşıyan tüplerin (safra kanallarının) iltihaplanması ve enfeksiyonunu ve safra kesesi ve bağırsaklara (kolanjit) ana damarının yüksek kan basıncını içerir. karaciğer (portal hipertansiyon).
Portal hipertansiyon, yemek borusu, mide ve bağırsakların damarlarında (varislerde) artan baskı ve şişmeye (dikkat) neden olabilir. Bu damarlar yırtılabilir ve potansiyel olarak hayati tehlike yaratan gastrointestinal kanamaya (varis kanaması) neden olabilir. Etkilenen çocuklar ilerleyici karaciğer fonksiyon bozukluğu ve yara izi (siroz) ve nihayetinde karaciğer yetmezliği yaşayabilir.
- ARPKD’li çocuklar, yetersiz böbrek fonksiyonu veya midenin genişlemiş böbrekler, karaciğer ve / veya dalakla sıkışması nedeniyle beslenme güçlüğü çekebilir. Beslenme zorlukları ve kronik böbrek yetmezliği, etkilenen bireylerde zayıf büyümeye katkıda bulunabilir. Bazı çocuklar idrar yolu enfeksiyonlarına yatkın olabilir ve su ve tuz dengesi ile ilgili problemleri olabilir.
- ARPKD’li bazı çocuklar dalağın genişlemesini yaşayabilir (splenomegali). Splenomegali potansiyel olarak düşük seviyelerde kırmızı kan hücreleri (anemi), trombositler (trombositopeni) ve beyaz kan hücreleri (lökopeni) ile sonuçlanabilir. Anemi yorgunluk, soluk cilt, düzensiz kalp atışı ve nefes darlığına neden olabilir. Trombositopeni kolay morarma, kesiklerden uzun süre kanama, spontan burun kanaması ve ciltte yüzeysel kanama ile sonuçlanabilir (peteşiler). Lökopeni vücudun enfeksiyon ve hastalıklarla savaşma yeteneğini azaltır.
Genetik Görülme Sıklığı
Polikistik böbrek hastalığı oldukça yaygın bir genetik hastalıktır. Amerika Birleşik Devletleri’nde yaklaşık 500.000 kişiyi etkiliyor. Hastalığın otozomal dominant formu otozomal resesif formdan çok daha yaygındır. Otozomal dominant polikistik böbrek hastalığı 500 ila 1.000 kişiden 1’ini etkilerken, otozomal resesif tip 20.000 ila 40.000 kişiden 1’inde görülür.
Hastaların çoğuna utero veya doğumda tanı konmasına rağmen, hafif vakalar ergenliğe veya yetişkinliğe kadar belirgin olmayabilir.
Teşhis Yöntemleri ve Tedavileri
Nedenleri
ARPKD, otozomal resesif bir özellik olarak kalıtılan PKHD1 geninin mutasyonlarından kaynaklanır . Genetik hastalıklar, baba ve anneden alınan kromozomlarda bulunan belirli bir gen için alellerin kombinasyonu ile belirlenir.
Resesif genetik bozukluklar, bir birey hastalık geninin anormal bir kopyasını her bir ebeveynden aldığında ortaya çıkar. Bir birey genin normal bir kopyasını ve bir mutasyonu olan birini alırsa, kişi hastalık için taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte yüzde 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte yüzde 50’dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak normal olma şansı yüzde 25’tir. Risk erkeklerde ve kadınlarda aynıdır.
Araştırmacılar çoğu tipik ARPKD vakasının tek bir gene, özellikle de PKHD1genine mutasyondan kaynaklandığını belirlemiştir . PKHD1 geni kromozom 6 (6q21.1) uzun kolu (q) üzerinde yer almaktadır. PKHD1 büyük bir gendir ve bu gene birçok farklı mutasyon ARPKD’ye neden olur.
Otozomal dominant polikistik böbrek hastalığı (ADPKD), böbreklerde kist oluşumu ile karakterize genetik bir hastalıktır. ADPKD dominant bir hastalık olduğundan, etkilenen bireylerin çoğunda ARPKD’den farklı olarak aile PKD öyküsü vardır. Böbreklerdeki kist oluşumunun neden olduğu semptomlar arasında yüksek tansiyon (hipertansiyon), son kaburga ile kalça arasında vücudun yanlarında ağrı (gögüs ağrısı), idrarda kan (hematüri) ve böbreklerin aşamalı olarak zayıf fonksiyonu bulunur ( böbrek yetmezliği). Hastaların en az yüzde 50’sinde ADPKD, sonunda diyaliz veya böbrek nakli olmak üzere renal replasman tedavisi gerektiren evre böbrek hastalığına son verir. ADPKD basit bir böbrek hastalığı değildir ve vücudun diğer organ sistemleri kistlerin ve diğer hastalıkların gelişmesinden potansiyel olarak etkilenebilir (çoklu sistem bozukluğu). Her insanda bulunan spesifik semptomlar, ilgili spesifik organ sistemlerine bağlıdır. Karaciğer, pankreas, omuriliği ve beyni kaplayan bir zar (araknoid membran) ve semen (seminal veziküller) parçası olan sıvı üreten erkek üreme sisteminin bezleri kistler gelişebilir. ADPKD’li bireylerde kalbi ve kan damarlarını (kardiyovasküler sistem) etkileyen anormallikler de görülebilir.
Çeşitli farklı bozuklukların bir özellik olarak kistik böbrekleri olabilir. Bu bozukluklara bazen polikistik böbreğin sendromik biçimleri de denebilir. Bu bozukluklar genellikle ek nörolojik, dijital, göz ve / veya diğer semptomlara ve bunları ARPKD’den ayıran fiziksel bulgulara sahiptir. Bu bozukluklar arasında Bardet-Biedl sendromu, Meckel sendromu, Joubert sendromu, Senior-Loken sendromu, nefronofthis ve oral-fasiyal-dijital sendrom bulunur.
Teşhis
ARPKD, doğumdan önce klinik bulgulara dayanarak şüphelenebilir (örneğin, elle hissedilen göbek kitlesi, az gelişmiş akciğerler, oligohidramnios ve hipertansiyon). ARPKD teşhisi için sonogram, ultrason ve manyetik rezonans görüntüleme (MRG) içeren radyolojik görüntüleme kullanılabilir. Böbrek anormalliklerinin tespitine ek olarak, karaciğerdeki intraheptik kanalların tıkayıcı olmayan genişlemesini (dilatasyonu) tanımlamak için çeşitli radyolojik görüntüleme teknikleri de kullanılabilir.
Prenatal ultrason, genişlemiş böbrekleri ortaya çıkarabilir (bazı durumlarda doğumdan sonraki 18 hafta gibi). Bir ultrason ayrıca, gerici toplama tüpleri olan sayısız küçük kisti de açığa çıkarabilir. Gerçek böbrek kistleri de mevcut olabilir. Bununla birlikte, bir ultrason böbrek büyümesini veya oligohidramniosu tespit etmede başarısız olabilir.
PKHD1 genlerindeki mutasyonlar için genetik testler birçok farklı laboratuarda mevcuttur.
ARPKD tanısı konmuş en az bir hamilelik geçirmiş risk altındaki bazı aileler için doğum öncesi veya doğum öncesi genetik tanı için genetik testler kullanılabilir.
Tedavi
ARPKD’nin tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Özel tedaviler, böbrek ve karaciğer fonksiyonunun korunmasına yöneliktir. Bebeklik döneminde solunum güçlüğü çeken birçok çocuk nefes almanıza yardımcı olmak için mekanik ventilasyon gerektirebilir. Nitrik oksit gibi ilaçlar, akciğerlere oksijen vermek (oksijenlemek) için yardımcı olabilir.
Şiddetli vakalarda, idrar üretimindeki (oligurya) azalmış veya idrar geçişi (anüri yok) yaşayan yenidoğanlarda yaşamın ilk birkaç günü periton diyalizi gerekebilir.
İlaçlar yüksek kan basıncını, özellikle de anjiyotensin dönüştürücü enzim (ACE) inhibitörlerini kontrol etmek ve yönetmek için kullanılabilir. Bazı kişilerde, yüksek tansiyon tedaviye dirençli olabilir (refrakter) ve birden fazla ilaç gerektirecek kadar şiddetli olabilir. Antibiyotikler idrar yolu enfeksiyonları veya kolanjit tedavisinde kullanılabilir.
Bazı çocuklar D vitamini, demir, bikarbonat ve sitrat gibi besin takviyelerine ihtiyaç duyabilir. Yeterli sıvı ve tuz takviyesi de gerekli olabilir. Beslenme zorlukları ve büyüme gecikmeleri nedeniyle, bazı çocuklar midede küçük bir cerrahi açıklıktan (gastrostomi) bir tüp veya burun içinden, özofagustan aşağı ve mideye (nazogastrik tüp) tüp yerleştirmeyi gerektirebilir. Bu tüpler temel besinleri doğrudan sağlamak için kullanılır. Ciddi durumlarda, büyüme hormonu tedavisi gerekli olabilir.
Böbreklerin artık çalışmadığı son dönem böbrek hastalığı olan kişiler, diyaliz veya böbrek nakli gerektirir. Diyaliz, bir makinenin, böbrek – atık ürünlerin kan dolaşımından süzülmesini, kan basıncını kontrol etmeyi ve potasyum gibi gerekli temel kimyasal seviyelerini korumaya yardım etmesini sağlamak için kullanılan bir prosedürdür. Son dönem böbrek hastalığı geri dönüşümsüz değildir, bu nedenle bireyler yaşam boyu diyaliz tedavisi veya böbrek nakli gerektirecektir. Böbrek fonksiyon bozukluğunun son dönem böbrek hastalığına ilerleme hızı, bir kişiden diğerine büyük ölçüde değişebilir. Bazı bireyler çocukluk döneminde böbrek nakli gerektirir; diğerleri yetişkinliğe kadar nakil gerektirmeyebilir veya hiç olmayabilir.
Progresif portal hipertansiyon, portal ven ile inferior vena cava arasında, vücudun kanının üçte ikisinden kan akıtan ana ven arasında bir bağlantı yapılan portakaval şantla tedavi gerektirebilir. Portal venin yüksek kan basıncını azaltmak için portacaval şant tasarlanmıştır.
Varis kanaması tıbbi bir acil durumdur ve acil tedavi gerektirir. Varis kanaması, sodyum klorid gibi bir çözeltinin etkilenen bir kan damarı içine enjekte edildiği bir prosedür olan skleroterapi ile tedavi edilebilir. Solüsyon kan damarını tahriş eder ve sonunda kanın pıhtılaşmasına neden olur. ARPKD’li bireylerin küçük bir yüzdesi sonunda karaciğer nakli gerektirebilir.
Erythropoietin, ARPKD’li bazı çocuklarda anemi yaşayan kırmızı kemik hücreleri üretmek için kemik iliğini stimüle etmek için kullanılabilir. Dalakların cerrahi olarak çıkarılması (splenektomi) bazı durumlarda şiddetli splenomegali tedavisinde kullanılır.
Genetik danışma, etkilenen bireyler ve aileleri için faydalı olabilir. Diğer tedavi semptomatik ve destekleyicidir.
Mesane veya böbrek enfeksiyonları. Böbrek hasarını önlemek için antibiyotik ile enfeksiyonların derhal tedavisi gereklidir.
PKD, tansiyonu kontrol etmek için yediklerinizde değişiklik gerektirebilir. Sağlıklı bir beslenme planının ardından kan basıncını düşürmeye yardımcı olabilir.
Doktorunuzun önerdiği Sağlıklı bir kiloyu koruyun. Sigara içilmemeli, içiliyorsa bırakılmalı.
Düzenli egzersiz. Haftanın çoğu günü en az 30 dakika orta derecede fiziksel aktivite,
Alkol kullanımını sınırlandırılmalı, gün boyunca bol miktarda sade su içmek, tüm içeceklerde kafeinden kaçınılması öneriler arasındadır.
Nisan 2018’de FDA, otozomal dominant polikistik böbrek hastalığının (ADPKD) tedavisi için “tolvaptan” adlı yeni bir ilacı onayladı. İlaç, bu tip PKD için risk altında olan yetişkinlerde böbrek fonksiyonlarının yavaşlamasını sağlamak için kullanılabilir.
Diğer kronik hastalıklarda olduğu gibi polikistik böbrek hastalığına sahip olmak bunaltıcı gelebilir. Arkadaşların ve ailenin desteği, kronik bir hastalıkla baş etmede önemlidir. Ek olarak, bir danışman, psikolog, psikiyatrist veya din adamı üye yardımcı olabilir.
Hastalığın Diğer İsimleri
- Polikistik Böbrek Hastalığı (PKD)
- Otozomal resesif polikistik böbrek hastalığı (ARPKD)
- Otozomal dominant polikistik böbrek hastalığı (ADPKD)
Kaynaklar
- https://ghr.nlm.nih.gov/condition/polycystic-kidney-disease#definition
- https://rarediseases.org/rare-diseases/autosomal-recessive-polycystic-kidney-disease/
- https://medlineplus.gov/kidneycysts.html
- https://www.niddk.nih.gov/health-information/kidney-disease/polycystic-kidney-disease
- https://www.mayoclinic.org/diseases-conditions/polycystic-kidney-disease/symptoms-causes/syc-20352820
- https://www.kidney.org/atoz/content/polycystic