Genel Bilgi
Tay-Sachs hastalığı, bir enzim eksikliğinin (hekzosaminidaz A), beyin ve sinir hücrelerinde gangliyozitler olarak bilinen bazı yağların (lipitler) aşırı birikmesine neden olduğu, nadir görülen, nörodejeneratif bir hastalıktır. Gangliosidlerin bu anormal birikimi, merkezi sinir sisteminin ilerleyici işlev bozukluğuna yol açar. Bu hastalık lizozomal depo hastalığı olarak sınıflandırılır. Lizozomlar hücrelerdeki ana sindirim üniteleridir. Lizozomlardaki enzimler, bazı kompleks karbonhidratlar ve yağlar dahil olmak üzere besin maddelerini parçalamaktadır veya “sindirmektedir”. Yağ gibi bazı maddeleri parçalamak için gerekli olan hekzosaminidaz A gibi bir enzim eksik veya etkisiz olduğunda, lizozomalde birikir. Buna “anormal depolama” denir. Lizozomda çok fazla yağ biriktiğinde hücreyi yok eden ve çevreleyen dokuya zarar veren toksik hale gelir. Tay-Sachs hastalığına bağlı semptomlar, ani seslere abartılı bir irkilme tepkisi, halsizlik, önceden edinilmiş becerilerin kaybı (yani psikomotor regresyon) ve ciddi şekilde azalmış kas tonusu (hipotoni) içerebilir. Hipotonili bebekler “disket” olarak tanımlanabilir. Hastalık ilerledikçe, etkilenen bebekler ve çocuklar gözlerin orta tabakasında kiraz kırmızısı lekeler, kademeli görme kaybı ve işitme kaybı, kas sertliği ve sınırlı hareketler (spastisite), nihai felç, kontrolsüz elektriksel rahatsızlıklar beyin (nöbet) ve bilişsel süreçlerin bozulması (demans). Tay-Sachs hastalığının klasik şekli bebeklik döneminde ortaya çıkar. Bu en yaygın şeklidir ve genellikle erken çocukluk döneminde ölümcüldür. Tay-Sachs hastalığının genç ve yetişkin formları da vardır, ancak bunlar nadirdir. Çocuk formuna sahip, alt form olarak da adlandırılan çocuklar, çocuk formuna sahip olanlardan daha sonra semptomlar geliştirir ve genellikle çocukluk veya ergenlikte yaşarlar. Geç başlangıçlı Tay-Sachs hastalığı olarak da adlandırılan yetişkin formu, ergenlik döneminden 30’lu yaşların ortalarına kadar herhangi bir zamanda oluşabilir. Belirtiler ve ciddiyet bir kişiden diğerine değişebilir. Bazı insanlar çocuk ve yetişkin formları arasında düşebilir.
Tay-Sachs hastalığı, otozomal resesif bir şekilde kalıtsaldır. Hastalık, hekzosaminidaz A enziminin üretimini düzenleyen HEXA geni olarak bilinen bir genin değişmesinden (mutasyonlarından) kaynaklanır. HEXA geni, 15 kromozomunun (15q23-q24) uzun koluna (q) eşlenmiştir. Tay-Sachs hastalığının tedavisi yoktur ve tedavi, ortaya çıkan spesifik semptomları hafifletmeyi amaçlar. Tay-Sachs hastalığı için bir başka isim GM2 gangliosidosis tip 1’dir. Sandhoff hastalığı ve hekzosaminidaz aktivatörü eksikliği olarak adlandırılan, semptomlara dayanarak Tay-Sachs hastalığından ayırt edilemeyen ve sadece altta yatanları belirlemek için yapılan testlerle ayırt edilebilen diğer iki ilgili hastalık vardır. sebeb olmak. Bu iki rahatsızlık aynı zamanda heksosaminidaz aktivitesinin azalmasına neden olur, ancak farklı genlerdeki değişikliklerden kaynaklanır. Toplu olarak, bu üç hastalık GM2 gangliosidoses olarak bilinir.
Şekil.1. Tay-Sachs Hastalığı
Genetik Değişiklikler ve Etken Faktörler
Tay-Sachs hastalığına, hekzosaminidaz alt birim alfa (HEXA) genindeki bir değişiklik (mutasyon) neden olur. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir genin mutasyonu meydana geldiğinde, protein ürünü hatalı, yetersiz veya eksik olabilir. Protein fonksiyonlarına bağlı olarak, bu beyin de dahil olmak üzere vücudun birçok organ sistemini etkileyebilir. HEXA geni, heksosaminidaz A enziminin üretimini düzenler. Hastalığa sahip kişilerde HEXA geninin 80’den fazla farklı mutasyonu tanımlanmıştır. HEXA geninin iki mutasyona uğramış kopyalarının (homozigotlar) miras alınması, vücut hücrelerinde GM2-gangliosid olarak bilinen yağ maddesini (lipit) parçalamak için gerekli olan hekzosaminidaz A enziminin eksikliğine neden olur. GM2-gangliyositinin parçalanamaması, beyin ve sinir hücrelerinde anormal birikimine ve sonunda merkezi sinir sisteminin ilerleyici bozulmasına neden olur. İnfantil Tay-Sachs hastalığında, neredeyse tamamen heksosaminidaz A eksikliği vardır. Geç başlangıçlı Tay-Sachs hastalığında, heksosaminidaz A enzim aktivitesinde eksiklik vardır. Bazı enzim aktivitesi olduğundan, hastalık daha az şiddetlidir ve infantil Tay-Sachs hastalığından çok daha yavaş ilerler. Geç başlangıçlı Tay-Sachs hastalığında enzim aktivitesinin tam miktarı bir kişiden diğerine büyük farklılıklar gösterir. Sonuç olarak, başlangıç yaşı, şiddeti, spesifik semptomlar ve geç başlangıçlı Tay-Sachs hastalığının ilerleme hızı da bir kişiden diğerine büyük ölçüde değişmektedir. HEXA geninde Tay-Sachs hastalığına neden olan değişiklikler otozomal resesif bir şekilde kalıtsaldır. Çoğu genetik hastalık, biri babadan diğeri de anneden alınan bir genin iki kopyasının durumuna göre belirlenir. Resesif genetik bozukluklar, bir birey aynı özellik için anormal bir genin iki kopyasını her bir ebeveynden bir tane aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen miras alırsa, kişi hastalık için taşıyıcı olur, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem değiştirilmiş geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte% 50’dir. Bir çocuğun her iki ebeveynden normal gen alma şansı% 25’tir. Risk erkeklerde ve kadınlarda aynıdır. Araştırmacılar, Tay-Sachs hastalığı geninin, 15 (15q23-q24) kromozomunun uzun kolunda (q) bulunduğunu belirlemiştir. Kromozomlar, insan hücrelerinin çekirdeğinde bulunur ve her birey için genetik bilgiyi taşır. İnsan vücudu hücrelerinin normalde 46 kromozomu vardır. 1 ila 22 arasında numaralandırılmış insan kromozomlarının çiftlerine otosomlar denir ve cinsiyet kromozomları X ve Y olarak adlandırılır. Erkeklerde bir X ve bir Y kromozomu vardır ve dişilerde iki X kromozomu vardır. Her kromozomun “p” işaretli kısa bir kolu ve “q” işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, “kromozom 11p13”, kromozom 11’in kısa kolundaki bant 13’ü belirtir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
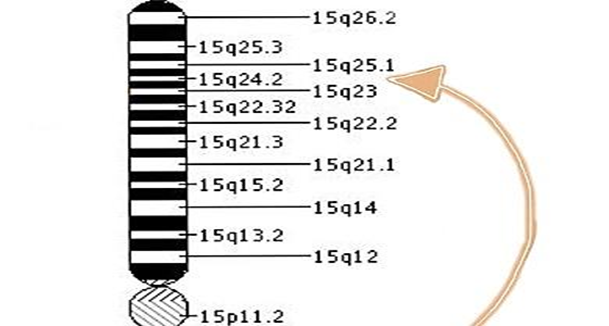
Şekil.2. Tay Sachs Hastalığı (İdeogram)
Belirti ve Semptomlar
Tay-Sachs hastalığının ilk belirtileri, bir kişinin (varsa) ne kadar beta-heksosaminidaz A enzim aktivitesine bağlı olarak bebeklikten yetişkinliğe kadar görülebilir. En yaygın biçimde, çocuk formunda, bebeklerde enzim aktivitesi yoktur veya çok düşük bir seviye (% 0.1’den az) bulunur. Genellikle yenidoğan döneminde sağlıklı görünürler, ancak 3 ila 6 ay içinde semptomlar geliştirirler. İlk belirti, gürültüye karşı abartılı bir ürkütücü tepki olabilir. Bu formdaki bebekler, yuvarlanma ve oturma (regresyon) gibi kilometre taşlarını kaybetmeye başlar ve yavaş yavaş felce neden olan kas zayıflığı geliştirir. Ayrıca zihinsel işlevlerini kaybederler ve çevrelerine karşı giderek daha tepkisiz hale gelirler. 12 aylıkken, daha hızlı bir şekilde bozulmaya başlar, körlük, tedavisi zor olan nöbetler ve yutkunma zorlaşır. Bu Tay-Sachs hastalığına sahip bebekler tipik olarak 4 yaşından sonra hayatta kalamazlar. En sık ölüm nedeni akciğer iltihabından (bronkopnömoni) gelen komplikasyonlardır. Çocuk formu daha az yaygındır ve tipik olarak normal aktivitenin% 1’inden daha az enzim aktivitesine sahip olması ile karakterize edilir. Tam olarak ne kadar aktivite olduğuna bağlı olarak, belirtiler çocukluk döneminde, en sık 2 ve 5 yaşları arasında herhangi bir zamanda başlayabilir. Bu forma sahip çocuklar sıklıkla sık enfeksiyonlar, davranışsal problemler geliştirir ve yavaş yavaş ilerleyen hareket kontrolü, konuşma ve Zihinsel işlev. Ayrıca nöbet geçirmeye başlayabilir ve görüşlerini kaybedebilirler. Çocuk formuna sahip çocuklar, geç çocukluk veya ergenlik döneminden vefat etmeden önce, genellikle birkaç yıl boyunca hiçbir yanıt veya farkındalığa sahip değillerdir. Enfeksiyon yaygın bir ölüm nedenidir. Bazen yetişkin veya kronik form olarak adlandırılan geç başlangıç formu da daha az yaygındır ve normal enzim aktivitesinin% 10’undan azına sahip olması ile karakterize edilir. Belirtiler ve ciddiyet, bu forma sahip kişiler arasında daha fazla değişiklik gösterir. Belirtiler çocuklukta yetişkinliğe kadar başlayabilir, ancak hastalık ergenliğe veya yetişkinliğe kadar sıklıkla teşhis edilmez. Nörolojik bozukluk yavaş yavaş ilericidir ve sakarlığa ve koordinasyon kaybına, kas güçsüzlüğü, titreme, konuşma zorluğu veya yutkunma ve kontrol edilemeyen kas spazmları ve hareketlerine neden olabilir. Pek çok insan sonunda mobilite yardımına ihtiyaç duyar. Bu formu olan bazı insanlarda ilk belirgin semptom şizofreni gibi ciddi bir psikiyatrik bozukluktur. Zarar görmüş zihin veya demans gelişebilir veya gelişmeyebilir. Geç başlangıç formuna sahip bazı kişiler hastalık nedeniyle kısaltılmış bir ömre sahiptir, diğerleri ise değildir.
Teşhis
Tay-Sachs hastalığının teşhisi, kapsamlı bir klinik değerlendirme ve vücuttaki heksosaminidaz A seviyelerini ölçen kan testleri gibi özel testlerle doğrulanabilir. Heksosaminidaz A, Tay-Sachs hastalığına sahip kişilerde azalır ve çocukluk formunda yoktur veya neredeyse yoktur. Moleküler genetik testler, Tay-Sachs hastalığının teşhisini doğrulayabilir. Moleküler genetik testler, hastalığa neden olduğu bilinen HEXA genindeki mutasyonları tespit edebilir, ancak sadece uzman laboratuvarlarda bir teşhis servisi olarak kullanılabilir. Bazı durumlarda, amniyosentez ve koryon villus örneklemesi (CVS) gibi özel testlere dayanarak doğumdan önce (doğum öncesi) Tay-Sachs hastalığının tanısından şüphelenilebilir. Amniyosentez sırasında, gelişmekte olan fetüsü çevreleyen bir sıvı numunesi çıkarılırken, CVS plasentadan bir bölümünden doku numunelerinin çıkarılmasını içerir. Bu örnekler, hekzosaminidaz A’nın mevcut olup olmadığını veya Tay-Sachs hastalığı olan kişilerde olduğu gibi, büyük ölçüde azalmış seviyelerde bulunup bulunmadığını belirlemek için incelenmiştir. Buna enzim deneyi denir. Doğum öncesi tanı, eğer HEXA geninde spesifik hastalığa neden olan mutasyon ailede bilinirse, CVS veya amniyosentez yoluyla elde edilen doku örneklerinin moleküler genetik testleriyle de mümkündür. Kan testleri, bireylerin Tay-Sachs hastalığı için taşıyıcı olup olmadığını belirleyebilir (yani, hastalık geninin bir kopyasına sahiptir). Tay-Sachs hastalığı olan bireylerin akrabaları, hastalık geninin taşıyıcı olup olmadıklarını belirlemek için test edilmelidir. Bir çocuk sahibi olmayı ve herhangi bir Yahudi soyuna sahip olmayı planlayan çiftlerin (sadece Aşkenazi değil) hamilelikten önce taşıyıcı taramasından geçmeleri teşvik edilir.
Tedavi
Tay-Sachs hastalığının spesifik bir tedavisi yoktur. Tedavi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk doktorları, nörologlar, konuşma patologları, işitme problemlerini gören ve tedavi eden uzmanlar (odyologlar), göz uzmanları ve diğer sağlık profesyonellerinin etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir. Genetik danışma, etkilenen bireyler ve aileleri için faydalı olabilir. Tüm aile için psikososyal destek önerilmektedir. Beslenme zorluğu potansiyeli nedeniyle, bebeklerin beslenme durumu ve uygun hidrasyon için izlenmesi gerekir. Beslenme desteği ve takviyesi gerekli olabilir ve bazen bir besleme tüpünün yerleştirilmesi gerekebilir. Beslenme desteğine ek olarak, yiyecek, sıvı veya diğer yabancı maddelerin yanlışlıkla akciğerlere (aspirasyon) girmesini önlemek için bir besleme tüpü gerekebilir. Antikonvülsanlar, Tay-Sachs hastalığına sahip bazı kişilerde ortaya çıkan nöbetleri tedavi etmek için kullanılabilir, ancak her insanda etkili olmayabilir. Ayrıca, nöbetlerin tipi ve sıklığı, bir kişinin ilaç tipinde veya dozajında değişiklik gerektirecek şekilde değişebilir.
Sıklık
Tay-Sachs hastalığı genel popülasyonda çok nadir görülür. Bu hastalığa neden olan genetik mutasyonlar Aşkenazi (doğu ve orta Avrupa) Yahudi mirası insanlarında diğer kökenden daha yaygındır. Bu hastalıktan sorumlu mutasyonlar, Quebec’in bazı Fransız-Kanadalı topluluklarında, Pennsylvania’daki Eski Düzenli Amish topluluğunda ve Louisiana’nın Cajun popülasyonunda da daha yaygındır.
Kalıtım Paterni/Deseni
Bu durum otozomal resesif bir kalıtsal kalıtımla ifade edilir, yani her bir hücredeki genin her iki kopyası da mutasyonlara sahiptir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve semptomlarını göstermezler.
Hastalığın Diğer Adları
GM2 gangliosidosis, tip 1
HexA eksikliği
B varyantı GM2 gangliosidozu
Heksosaminidaz A eksikliği
Heksosaminidaz alfa-alt birim eksikliği (değişken B)
Sfingolipidoz, Tay-Sachs
TSD
Gangliosidosis GM2, tip 1
Kaynak
[1] https://rarediseases.info.nih.gov/diseases/7737/tay-sachs-disease
[2] https://ghr.nlm.nih.gov/condition/tay-sachs-disease
[3] https://rarediseases.org/rare-diseases/tay-sachs-disease/
Görsel Kaynak