(Xeroderma pigmentoso)
Genel Bilgi
De Sanctis-Cacchione sendromu, nörolojik anormallikler, zihinsel gerilik, alışılmadık kısa boy (cücelik) ve testislerin veya yumurtalıkların az gelişmişliği ile bağlantılı olarak ortaya çıkan xeroderma pigmentosum’un (XP) cilt ve göz semptomları ile karakterize oldukça nadir görülen bir hastalıktır. . Xeroderma pigmentosum, ultraviyole ışığına (ışığa duyarlılık), cilt rengindeki renk bozukluklarına ve çeşitli göz bozuklukları ve cilt kanserlerinin olası gelişimine karşı yüksek reaksiyon gösteren, nadir görülen kalıtsal cilt hastalıkları grubudur. De Sanctis-Cacchione sendromu ile ilişkili en yaygın nörolojik anormallikler düşük zeka, anormal derecede küçük bir kafa (mikrosefali), gönüllü hareketi koordine etme kabiliyetinin kaybı (ataksi) ve / veya yok (refleksi) veya zayıflamış (hiporefleksi) refleksleridir.
Orpha.net web sitesinde “Bu hastalık Xeroderma pigmentosum’a taşındı” bilgisi karşımıza çıkmaktadır. De Sanctis-Cacchione sendromu, başlangıçta ağır nörolojik anormallikleri olan XP vakalarına atfedilen ancak genel kullanımda olmayan bir terimdir.
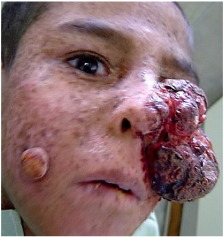
Görsel 1 kaynak: https://www.sciencedirect.com/science/article/pii/S2352647515000374
De Santis cacchione sendrome
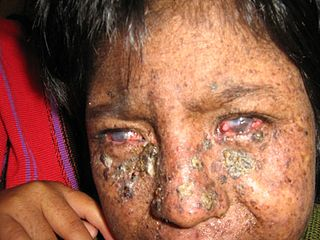
Görsel 2, kaynak: http://flipper.diff.org/app/items/info/6206
Genetik Değişiklikler/ Etken Faktörler
De Sanctis-Cacchione sendromunun belirtileri, vücudun genlerin yapı taşlarına (DNA) zarar verememesi nedeniyle ortaya çıkar. Hasar, güneş ışınları gibi ultraviyole ışığa maruz kalmasından kaynaklanır. Herkesin bu hasarı karmaşık bir işlemle onarabilen bazı bağ dokusu hücreleri (fibroblastlar) vardır. Bununla birlikte, De Sanctis-Cacchione sendromundan etkilenen insanlarda bulunan fibroblastlar, DNA’larını onarma yeteneğinden yoksundur veya kapasiteleri azalır. Ek olarak, etkilenen bazı kişilerin hücreleri güneş ışığına zarar veren cildi düzgün şekilde onaramaz.
Vücudun güneş ışığına zarar veren DNA’yı onarma kapasitesine bağlı olarak çeşitli XP formları (alt bölümler) tanımlanmıştır. XP’nin alt bölümlerinden herhangi biri De Sanctis-Cacchione sendromunda ortaya çıkabilir. Bununla birlikte klasik XP formu (xeroderma pigmentosum, Stype A) veya xeroderma pigmentosum, D tipi en sık De Sanctis-Cacchione sendromu ile birlikte bulunur.
Araştırmacılar, bazı De Sanctis-Cacchione sendromu vakalarının, kromozom 10’un (10q11) uzun kolunda (q) bulunan belirli bir genin bozulmasından veya değişmesinden kaynaklanabileceğini belirlemiştir. İnsan hücrelerinin çekirdeğinde bulunan kromozomlar her bireyin genetik bilgisini taşır. İnsan kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve erkeklerde bir X ve bir Y kromozomu ve kadınlarda iki X kromozomu içeren ek bir 23. cinsiyet kromozomu çiftidir. Her kromozomun “p” işaretli kısa bir kolu ve “q” işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, “kromozom 10q11”, kromozom 10’un uzun kolundaki bant 11 ile ilgilidir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
Belirti ve Semptomlar
De Sanctis-Cacchione sendromunda en erken semptomlar, ultraviyole ışığına maruz kaldıktan sonra meydana gelen aşırı çil ve kabarma dahil xeroderma pigmentosum (XP) ile ilişkili cilt anormallikleridir (ışığa duyarlılık). Bazı durumlarda, güneş ışığı ile temas ettikten hemen sonra ağrı ve kabarma meydana gelebilir. Akut güneş yanığı ve cildin kalıcı kızarıklığı veya iltihabı (eritem) de De Sanctis-Cacchione sendromunun erken belirtileridir. Çoğu XP vakasında, bu semptomlar doğumdan hemen sonra veya üç yaşından sonra görülebilir. Bununla birlikte, bazı nadir durumlarda, daha sonra çocukluk döneminde semptomlar belirgin olmayabilir. De Sanctis-Cacchione sendromunun çoğu vakasında, başlangıç genellikle bebeklik döneminde görülür.
Sıklıkla De Sanctis-Cacchione sendromu ile ilişkili ek cilt semptomları, cildin alışılmadık derecede koyu (hiperpigmentasyon) veya açık (hipopigmentasyon) alanlarını içerir. Bazı durumlarda, cilt renginin tamamen kaybolması (depigmentasyon) ve / veya aşırı skar oluşabilir. Siğil benzeri lezyonlar (aktinik keratozlar) yanı sıra, cildin yüzeyine yakın küçük kan damarlarının anormal şekilde genişlemesinden kaynaklanan küçük kırmızı cilt lezyonları (telangiektaziler) gelişebilir. Cilt ayrıca zayıflayabilir ve kolayca zarar görebilir. Dejeneratif (atrofik) değişiklikler meydana gelebilir ve cilt kuru ve pürüzsüz görünebilir.
De Sanctis-Cacchione sendromu olan çoğu çocuğun genellikle bir veya daha fazla nörolojik anomalisi vardır; en sık görülen zeka düşüktür. Diğer anormallikler, alışılmadık derecede küçük bir baş (mikrosefali); işitme bozukluğu (duyusal sağırlık); eksik (arefleksi) veya zayıflamış (hiporefleksi) refleksleri; ve / veya bazı kaslarda sertlik ve hareket kısıtlılığına (spastisite) neden olan sertliğin artması. Etkilenen bireyler ayrıca gönüllü hareketleri (ataksi) ve / veya vücudun anormal istemsiz hareketlerini, kontrolsüz sarsıntılı hareketler (anormal yavaş hareketler (koreotetoz)) ile birlikte koordine etme kabiliyetlerini de gösterebilir.
De Sanctis-Cacchione sendromu, zaman zaman, serebellum (serebellar atropi) olarak bilinen beynin bir kısmının yavaş ilerleyici dejenerasyonu ve / veya beynin diğer kısımlarının (yani, korteks, baz pontisi) degrasyonunu içeren bir grup ilerleyici bozukluktan herhangi biriyle ilişkilidir. ve aşağı olivary çekirdekleri. Bu klinik tablo, kalıtsal olivopontocerebellar atrofilerde görülene benzer. Belirtiler, bozuk kas koordinasyonu (ataksi), titreme, istemsiz hareketler ve konuşma bozukluklarını (dizartri) içerebilir. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veri Tabanında arama teriminiz olarak “Kalıtsal Olivopontocerebellar Atrofi” yi seçin.)
De Sanctis-Cacchione sendromu olan bireyler ayrıca alışılmadık derecede yavaş gelişme, kısa boylanma (cücelik), zihinsel gerilik ve / veya testis veya yumurtalıkların (hipogonadizm) yetersiz fonksiyonu ile sonuçlanan derin büyüme gecikmeleri sergileyecektir.
Benign cilt tümörleri, De Sanctis-Cacchione sendromu ile ilişkili olabilir ve beş yaşından önce başlaması mümkündür. Bunlar, kan damarlarından (anjiyomlar) oluşan tümörler ve / veya sıklıkla cildin güneşe maruz kalan bölgelerinde ortaya çıkan hızla büyüyen tümörler gibi, önceden malign veya iyi huylu (kanserli olmayan) tümörleri içerebilir.
De Sanctis-Cacchione sendromu olan kişiler erken yaşta cilt kanseri yaşarlar. Örneğin, malign melanom, bazal hücreli karsinom ve skuamöz hücreli karsinom gibi cilt kanserleri bu rahatsızlığı olan kişilerde sıklıkla görülür; En sık etkilenen bölgeler baş, boyun ve yüzdür. (Bu bozukluklar hakkında daha fazla bilgi için bu raporun İlgili Bozuklukları bölümüne bakın.)
De Sanctis-Cacchione sendromunda, XP ile ilişkili göz semptomlarından bazıları mevcut olabilir. Bunlar ışığa aşırı derecede toleranssızlık içerebilir (fotofobi); gözlerin kornealarının iltihabı (keratit); gözlerin beyaz kısmını kaplayan zarı iltihabı (konjonktivit); göz kapaklarının dışa dönük (ektropion); ve / veya göz kapaklarının içe dönük (entropion). Cilt ve gözlerle ilgili semptomların şiddeti, ultraviyole ışığına maruz kalma miktarına bağlı olabilir.
Genetik Görğlme Sıklığı
De Sanctis-Cacchione sendromu, otozomal resesif bir özellik olarak kalıtsaldır. Klasik genetik hastalıklar da dahil olmak üzere insan özellikleri, biri babadan diğeri de anneden alınan iki genin etkileşiminin ürünüdür.
Resesif genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı anormal geni aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte% 50’dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak normal olma şansı% 25’tir.
Belgelenen vakaların yaklaşık yüzde 30’unda, De Sanctis-Cacchione sendromundan etkilenen bireylerin, kanla ilişkili (akraba) anne-babaları olmuştur. Tüm bireyler 4-5 anormal gen taşır. Yakın akraba olan ebeveynlerin (akraba), ilişkisiz ebeveynlerden her ikisinin de aynı anormal geni taşıması olasılığı daha yüksektir, bu da resesif genetik bozukluğu olan çocukların görülme riskini artırır.
ABD ve Avrupa’da tahmini yaygınlığı 1 / 1.000.000 olup, diğer ülkelerdeki (örneğin Japonya, Kuzey Afrika ve Pakistan), özellikle de yüksek derecede akrabalık dereceli topluluklarda daha yüksek rakamlara sahiptir.
Kalıtım Paterni Deseni
De Sanctis-Cacchione sendromu erkekleri ve kadınları eşit sayıda etkileyen çok nadir görülen bir hastalıktır. Batı tıp literatüründe yaklaşık 200 vaka bildirilmiş olmasına rağmen, bu hastalığın kesin vakalarının sayısı bilinmemektedir. Semptomların başlangıcı genellikle yaşamın ilk yılında ortaya çıkar, ancak nadir durumlarda erken veya geç çocukluk döneminde ortaya çıkabilir. Bazı nörolojik semptomların başlangıcı beş ila 10 yaşları arasında veya hatta yaşamın ikinci on yılında ortaya çıkabilir. De Sanctis-Cacchione sendromu ilk olarak 1932 yılında tıp literatüründe tanımlanmıştır.
XP, DNA onarımında rol alan 8 gendeki mutasyonlardan kaynaklanır. Bu genlerin yedi, DA için XPG ( ERCC5 ), nükleotid onarım (EŞ) yer alırlar. XPV veya POLH , UV kaynaklı hasarı içeren DNA’yı kopyalamak için gereken DNA polimeraz etasını kodlar.
Teşhis Yöntemleri ve Tedavileri
Teşhis
De Sanctis-Cacchione sendromunun teşhisi, xeroderma pigmentosum’un bir veya daha fazla nörolojik anormallik, zihinsel gerilik, cücelik ve testislerin veya yumurtalıkların yetersiz fonksiyonu ile birlikte ortaya çıktığı ortaya çıktığında doğrulanabilir.
Doğumdan önce (doğum öncesi) XP tanısı, amniyosentez adı verilen özel bir prosedür kullanılarak doğrulanabilir. Bu prosedür sırasında, fetüsü çevreleyen bir sıvı örneği alınır ve fetüsün XP olup olmadığını belirlemek için testler yapılır. Bu prosedür genellikle XP öyküsü olan aileler için bir tarama sürecinin bir parçası olarak yapılır.
De Sanctis-Cacchione sendromunun diğer semptomlarının mevcut olup olmadığını belirlemek için kapsamlı bir klinik değerlendirme yapılmalıdır (yani nörolojik anormallikler, zihinsel gerilik, cücelik ve hipogonadizm. Bu değerlendirme, manyetik rezonans görüntüleme (MRI) gibi nörogörüntüleme çalışmalarını içerebilir. ve bilgisayarlı tomografi (BT) taramaları.
De Sanctis-Cacchione sendromu tanısını doğrulamak için özel laboratuvar testleri kullanılabilir. Bu testler, beyaz kan hücrelerinde (lenfositler), karaciğer hücrelerinde, kornea hücrelerinde ve De Sanctis-Cacchione sendromundan etkilenen insanlardan alınan cilt hücrelerinde hatalı DNA onarımını tespit edebilir. Bu tür testler sırasında, hücreler UV radyasyonuna ve / veya bazı kanser üreten maddelere (kanserojenler) maruz kalırlar. Bu maddelere maruz kaldıktan sonra, bu hücrelerin kusurlu DNA onarım işlemi belirginleşir.
Tedavi
De Sanctis-Cacchione sendromu olan bireylerde, cilt lezyonlarının ve diğer komplikasyonların (örneğin, cilt kanserleri ve bazı nörolojik semptomlar) gelişimini önlemek için cildin güneş ışığından (örn. Topikal güneşten koruyucular, güneş gözlükleri, çift kat giysi) tamamen korunması gerekir. . De Sanctis-Cacchione Sendromu olan etkilenen bireyler, ultraviyole ışığa maruz kalmamak için gündüz saatlerinde dış mekan etkinliklerini sınırlandırmalıdır. Sigara dumanındakiler gibi kimyasal kanserojenlerden kaçınılması da önerilmektedir.
Deri kanseri olan kişiler için, lezyonların erken tespiti ve cerrahi olarak çıkarılması esastır. Derinin ve gözlerin uzmanlar tarafından düzenli muayenesi önerilir. Genetik danışmanlık, etkilenen bireyler ve aileleri için faydalı olacaktır. Diğer tedavi semptomatik ve destekleyicidir.
XP için bir tedavi yoktur, ancak herhangi bir cilt kanserini değerlendirmek ve tedavi etmek için güneşten korunma ve düzenli takip yaşam süresini uzatır. Nörolojik hastalığı ve titiz UV korumasına sahip olmayanlar için prognoz iyidir. Bununla birlikte, nörolojik anormallikler ilericidir ve ömrünün kısalmasına neden olabilir.
Araştırma Terapileri
De Sanctis-Cacchione sendromunu tedavi etmek için kozmetik cerrahi prosedür (dermabrazyon), kimyasal peeling, tümör eksizyonu ve / veya yüz cilt grefti kullanılmıştır. Dermabrazyon ve topikal 5-flüoroürasil, premalign veya erken cilt lezyonlarının tedavisinde etkili olabilir. Bir katalaz kreminin uygulanması bazı çocuklarda tümörleri önlüyor gibi görünmektedir. A vitamini türevlerini içeren merhemler veya kremler de incelenmektedir.
Kapsüllenmiş T4 endonükleaz VB lipozomu (T4N5), cilt kanserlerini ve xeroderma pigmentosum ile ilişkili diğer cilt anormalliklerini önlemek için kullanılan bir yetim ilaçtır. İlk çalışmalar, topikal T4N5 ile tedavinin, bazen De Sanctis-Cacchione sendromu ile bağlantılı cilt kanseri gelişim hızını yavaşlattığını göstermiştir. Xeroderma pigmentosum tedavisinde bu ilacın uzun vadeli güvenliğini ve etkinliğini belirlemek için daha fazla çalışmaya ihtiyaç vardır.
De Sanctis-Cacchione sendromu ile ilişkili göz anormalliklerinin tedavisi için çeşitli tedaviler incelenmektedir. Ameliyat, gözleri etkileyen problemler nedeniyle sıklıkla mümkün olmamakla birlikte, korneanın bir kısmının çıkarıldığı ve değiştirildiği (keratoplasti) özenle seçilmiş kişilerde çalışmalar yürütülmektedir. Yapay gözyaşı ve yumuşak kontakt lensler de kullanılmıştır.
Hastalıkla İlişkili Genler
Aşağıdaki bozuklukların belirtileri De Sanctis-Cacchione sendromununkilerle benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Cockayne sendromu, kısa boy ile karakterize nadir görülen kalıtsal bir hastalıktır; ayırt edici cilt anormallikleri; karakteristik kraniyofasiyal malformasyonlar ve erken yaşlanmış (progerik) bir görünüm; görme bozukluğu ile sonuçlanan göz anormallikleri; sağırlık; ve / veya zihinsel geriliği. Bozukluğu olan bireylerde, göz anormallikleri, göz sinirlerinin kademeli olarak bozulmasını (optik atrofi), gözleri kaplayan sinir bakımından zengin zarın ilerleyen dejenerasyonunu (retina dejenerasyonu) ve / veya göz merceklerinin anormal şekilde bulanıklaşmasını içerebilir. (katarakt). Ek olarak, cilt güneş ışığına karşı anormal derecede hassas olabilir; bu da yara izi, pullu döküntü, anormal pigmentasyon ve etkilenen alanların dejenerasyonu (atrofi) ile sonuçlanabilir. Cockayne sendromu olan bireyler ayrıca erken burun görünümüne ve ince burun, batık gözler ve / veya çıkıntılı alt çene (prognatizm) dahil olmak üzere karakteristik yüz anormalliklerine sahip olabilir. Etkilenen bireyler, aynı zamanda nadir görülen saçı ve çeşitli iskelet malformasyonlarını içeren ek fiziksel anormalliklere de sahip olabilir. Cockayne sendromu, otozomal resesif bir özellik olarak kalıtsaldır. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “Cockayne” ı seçin.)
Pigmentli kserodermoid, kseroderma pigmentozuma benzer semptomların geç başlangıcı ile karakterize bir durumdur. Bu belirtiler güneş ışığına yüksek reaksiyon gösterebilir (ışığa duyarlılık); cilt renk değişikliği; küçük kırmızı cilt lezyonları (telangiektazi); göz anormallikleri; ve / veya iyi huylu (kanserli olmayan) tümörler (anjiyomlar ve keratoakanttomlar). Etkilenen bireyler bazı cilt kanseri türlerinin erken yaşlarında karşılaşabilirler (örneğin, bazal hücreli karsinom, skuamöz hücreli karsinom, malign melanom). Pigmentli xerodermoid olan kişiler ultraviyole ışıktan zarar gören DNA’yı onarabilir; Ancak, onarım sonrası işlem hatalı. Pigmentli kserodermoidin nedeni bilinmemektedir. Bununla birlikte, bazı bilim adamları, otozomal resesif bir özellik olarak kalıtsal olabileceğine inanmaktadır.
Aşağıdaki bozukluklar, ikincil özellikler olarak De Sanctis-Cacchione sendromu ile ilişkilendirilebilir. Ayırıcı tanı için gerekli değildir:
Malign melanom, cildin üst tabakasının melanin hücrelerinden (epidermis) veya mollerde (nevi) bulunabilen benzer hücrelerden gelişen bir cilt kanseri türüdür. Erken evrelerde, çoğu melanom herhangi bir spesifik semptom üretmez. Daha sonra iyileşmeyen lezyonlar veya boyut veya renkte değişiklikler gösteren mevcut moller olarak görünebilirler. Bu tip cilt kanseri nihayetinde düşük cilt seviyelerine ve bitişik dokulara yayılır ve vücudun hayati organlarında yeni tümör büyümelerine neden olabilir. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “Melanom” seçeneğini seçin.)
Bazal hücreli karsinomlar cilt yüzeyinde ortaya çıkan tümörlerdir. Küçük, parlak, sert doku kütleleri (nodüller); yassı, yara benzeri lezyonlar (plaklar); veya kalın, kuru, gümüş pullarla kaplı kırmızı lekeler. Eğer tedavi edilmezse, bazal hücreli karsinom vücudun diğer bölgelerine yayılabilir (metastaz yapmaz). Bu tip cilt kanseri, biyopsi olmadan sedef hastalığından veya lokalize dermatitten ayırt edilmesi zor olabilir. Güneş ışığına veya iyonlaştırıcı radyasyona aşırı maruz kalmak bu tür cilt kanserine neden olabilir.
Skuamöz hücreli karsinomlar, genellikle cildin güneşe maruz kalan bölgelerinde ortaya çıkan, ancak vücudun herhangi bir yerinde görülebilen tümörlerdir. Bu cilt kanseri biçimi, pullu veya kabuklu bir yüzeye sahip görünen lezyonlarla karakterizedir. Bu lezyonlar altta yatan dokulara yayılabilir; Bununla birlikte, uygun tedavi ile, Squamous hücreli karsinom genellikle tedavi edilebilir. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “Squamous Cell Carcinoma” seçeneğini seçin.
Hastalığın Diğer İsimleri
- Xerodermic Idiocy
- Xeroderma pigmentoso
Kaynaklar