Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
KGB sendromu ilk 1975 yılında tanımlanıyor. Bu sendromun ismi ise Dr Optiz tarafından bu sendromu takip ettiği ailelerin soyisim ilk harflerinden gelmektedir.(1)
KBG sendromu, üst merkezi kesici dişlerin makrodontisi, ayırt edici kraniyofasiyal bulgular, kısa boy, iskelet anomalileri ve global gelişimsel gecikmeyi, nöbetleri ve zihinsel engeli içeren nörolojik tutulumla karakterizedir. KBG sendromunun yetersiz teşhis edilmesinin muhtemel olduğunu, çünkü zihinsel engellilik de dahil olmak üzere birçok özelliğin ılımlı olduğunu ve hiçbir özelliğin teşhis için önkoşul olmadığını belirtiliyor (2).
Belirti ve Semptomlar
KBG sendromunun karakteristik bir özelliği alışılmadık derecede büyük üst ön dişlerdir (makrodonti). Diğer belirgin yüz özellikleri arasında geniş, kısa bir kafatası ( brakisefali), bir üçgen yüz şekli, geniş aralıklı gözler (hipertelorizm), ortada birlikte büyüyebilecek geniş kaşlar (synophrys), belirgin bir burun köprüsü, burun ve üst dudak arasında uzun bir boşluk (uzun filtrum) ve ince üst dudak bulunur.
KBG sendromu olan kişilerde yaygın bir iskelet anomalisi olarak kemiklerin mineralizasyonu yavaşlamıştır (gecikmiş kemik yaşı); örneğin, etkilenen 3 yaşındaki bir çocuğun 2 yaşında tipik bir kemiği olabilir. Ayrıca, etkilenen bireylerin omurgalarında (omur) ve kaburga kemiklerinde anormallikler olabilir. Ayrıca, olağandışı kısa ya da kavisli beşinci (pembemsi) parmaklar ( brakidaktili ) dahil olmak üzere, ellerin veya ayakların kemiklerinde anormallikler olabilir. (klinodaktili, düz ayaklar ( pes planus)).
KBG sendromunda zihinsel ve hareket kabiliyetlerinin gelişimi de gecikmektedir. Etkilenen bireylerin çoğu konuşmayı ve yürümeyi normalden daha geç öğrenir ve zihinsel yetersizliği hafif ila orta şiddettedir. Bu hastalığı olan çoğu insan, hiperaktivite gibi davranışsal veya duygusal problemlere sahiptir; anksiyete; veya bozulmuş iletişim ve sosyal etkileşimlerle karakterize otizm spektrum bozukluğu da ayrıca incelenmelidir.
KBG sendromunun daha az yaygın özellikleri ise işitme kaybı, nöbetler ve kalp kusurlarını içerir.
Görsellerle belirtiler:
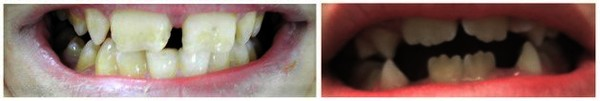
makrodonti (https://ghr.nlm.nih.gov/art/large/kbgs-teeth.jpeg)
{kind=link}
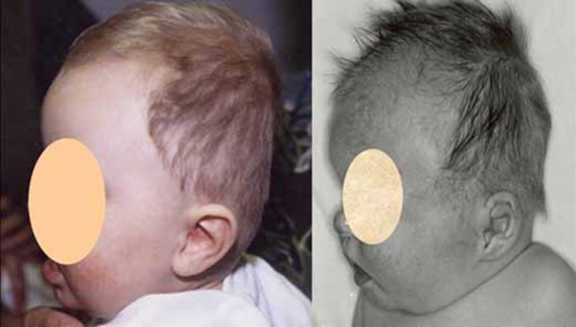
brakisefali (https://ghr.nlm.nih.gov/art/large/brachycephaly.jpeg)
{kind=link}
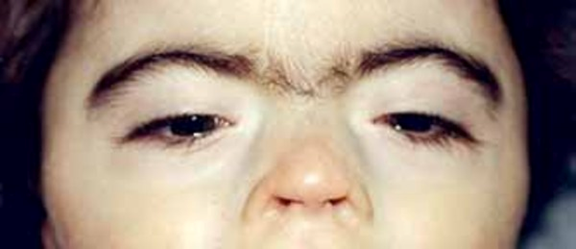
kaşların devamlılığı synophrys(https://ghr.nlm.nih.gov/art/large/synophrys.jpeg)
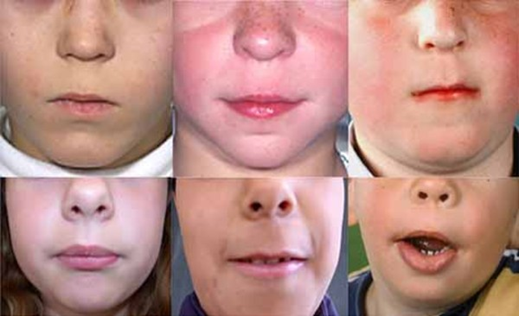
Uzun filtrum (https://ghr.nlm.nih.gov/art/large/long-philtrum.jpeg)
{kind=link}
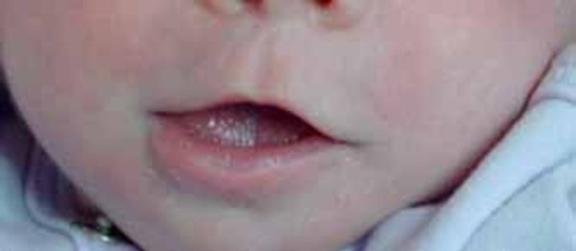
İnce üst dudak (https://ghr.nlm.nih.gov/art/large/thin-upper-lip.jpeg)
{kind=link}

Barkidaktili (https://ghr.nlm.nih.gov/art/large/brachydactyly-hand.jpeg)
{kind=link}
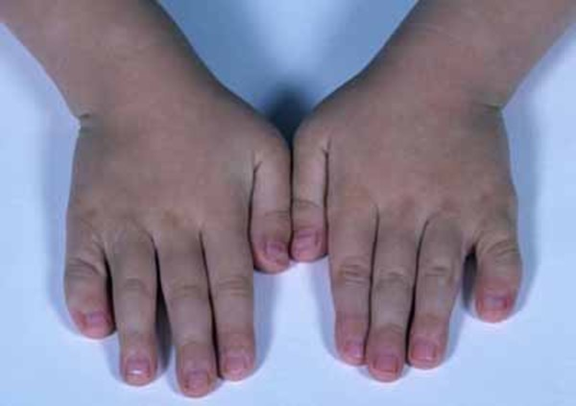
Klinodaktili (https://ghr.nlm.nih.gov/art/large/clinodactyly.jpeg)
{kind=link}
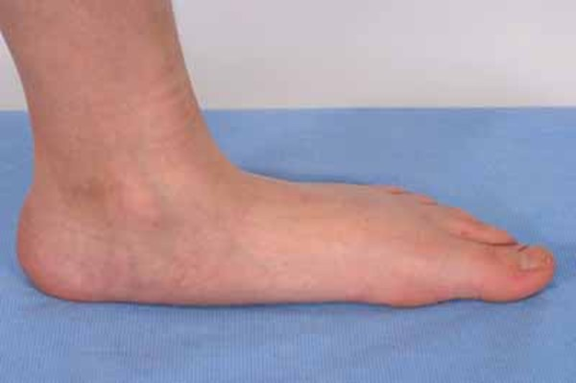
Pes planus veya düz taban (https://ghr.nlm.nih.gov/art/large/flat-feet1.jpeg)
{kind=link}
Genetik Görülme Sıklığı
KBG sendromu oldukça nadir görülen bir sendromdur. 2017 yılında tüm dünyada toplamda 200’den az hastaya tanı konmuştur. Kaynak: http://nebula.wsimg.com/8f16bb79bd36dd4c9cacdc494acdcc50?AccessKeyId=F45FA02690DBBB1D6A96&disposition=0&alloworigin=1
Kalıtım Paterni/Deseni
Tekin vd. (2004) tarafından bildirilen Anadolu’dan bir ailede; baba ve 2 oğlu KBG sendromu tanısı aldı ve otozomal dominant kalıtım olduğu genetik testler ile doğrulandı.(3) Fakat yine 2004 yılında yayınlanan bir başka makalede ise Maegawa ve diğ. (2004) hafif derecede etkilenen bir anneyi ve ağır şekilde etkilenen oğlunu bildirmiştir. Erkeklerin kadınlardan daha ciddi bir şekilde etkilendiği önceki raporlara dayanarak (örneğin, Parloir ve diğerleri, 1977 (4) ), bazı durumlarda kalıtımın X’e bağlı olabileceğini öne sürülmüştür (5).
Teşhis Yöntemleri ve Tedaviler
Teşhis;Tam bir klinik değerlendirme, ayrıntılı bir hasta ve aile öyküsü ve karakteristik fiziksel bulguların tanımlanmasından sonra KBG sendromu tanısından şüphelenilebilir. Teşhis ayrıca gen paneli analizi veya zihinsel yetersizliğin çoklu genetik nedenlerinin aynı anda araştırıldığı yeni nesil dizilim teknikleri ile de yapılabilir.
Tedavi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk doktorları, ortopedistler, ortopedi cerrahları, nörologlar, fizyoterapistler, konuşma terapistleri, ortodontistler ve diğer sağlık uzmanlarının etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir.
Genetik danışmanlık, etkilenen bireyler ve aileleri için önerilmektedir. Tüm aileye psikososyal destek de faydalı olabilir.
Ortopedik cerrahi, etkilenen kişilerin kalça ve omurga anormalliklerini düzeltmek için özellikle yardımcı olabilir. İşitme cihazları, konuşma terapisi ve kapsamlı diş bakımı da faydalı olabilir.
Hastalıkla İlişkili Genler
KGB sendromuna ANKRD11 genindeki bir değişiklik(mutasyon) veya ANKRD11 genini içeren 16q kromozomundan genetik materyal kaybı neden olur. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir genin mutasyonu meydana geldiğinde, protein ürünü hatalı, yetersiz veya eksik olabilir. Belirli bir proteinin fonksiyonlarına bağlı olarak, bu vücudun birçok organ sistemini etkileyebilir.
ANKRD11 gen sinir hücreleri (nöronlar) aktif olan bir proteini oluşturmak için talimatlar içerir. Bu proteinin tam rolü tam olarak anlaşılmamıştır. ANKRD11 değiştirildiğinde veya kaybolduğunda, bireyler bu proteinin yeterince işlevsel kopyalarını üretemezler. ANKRD11 geninin protein ürününün düşük seviyelerinin KBG sendromu semptomlarına neden olmasına karar vermek için daha fazla araştırma gereklidir .
KBG sendromu otozomal dominant paternde kalıtsaldır(3). Çoğu genetik hastalık, biri babadan diğeri de anneden alınan bir genin iki kopyasının durumuna göre belirlenir. Baskın genetik bozukluklar, belirli bir hastalığa neden olmak için değiştirilmiş veya eksik bir genin sadece bir kopyasının gerekli olduğu durumlarda ortaya çıkar. Etkilenen gen, her iki ebeveynden miras alınabilir veya etkilenen bireyde yeni, kendiliğinden bir mutasyonun (gen değişimi) sonucu olabilir. Buna “de novo” değişikliği denebilir. Değiştirilen geni veya eksik kromozom segmentini etkilenen bir ebeveynden yavrulara geçirme riski her hamilelik için% 50’dir. Risk erkeklerde ve kadınlarda aynıdır.
Hastalığın Diğer İsimleri
Diğer isimleri diyemesek de araştırmalarda şu şekilde de karşımıza çıkabilir:
- Makrodonti, zihinsel gerilik, karakteristik fasiyes, kısa boy ve iskelet anomalileri
- boy kısalığı-karakteristik fasiyes-zeka geriliği-makrodonti-iskelet anomalileri sendromu
- kısa boy, karakteristik fasiyes, makrodonti, zeka geriliği ve iskelet anomalileri
Kaynakça
1) Herrmann, J., Pallister, P. D., Tiddy, W., Opitz, J. M. The KBG syndrome–a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Art. Ser. XI(5): 7-18, 1975.
2) Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., Bademci, G., Agolini, E., Guo, S., Konuk, B., Kavaz, A., Blanton, S., Digilio, M. C., Dallapiccola, B., Young, J., Zuchner, S., Tekin, M. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 89: 289-294, 2011.
3) Tekin, M., Kavaz, A., Berberoglu, M., Fitoz, S., Ekim, M., Ocal, G., Akar, N. The KBG syndrome: confirmation of autosomal dominant inheritance and further delineation of the phenotype. Am. J. Med. Genet. 130A: 284-287, 2004.
4) Parloir, C., Fryns, J. P., Deroover, J., Lebas, E., Goffaux, P., van den Berghe, H. Short stature, craniofacial dysmorphism and dento-skeletal abnormalities in a large kindred: a variant of KBG syndrome or a new mental retardation syndrome. Clin. Genet. 12: 263-266, 1977.
5) Maegawa, G. H. B., Leite, J. C. L., Felix, T. M., da Silveira, H. L. D., da Silveira, H. E. Clinical variability in KBG syndrome: report of three unrelated families. Am. J. Med. Genet. 131A: 150-154, 2004.
Ayrıca temel bilgilerini kullandığım websiteleri:
1)https://ghr.nlm.nih.gov/condition/kbg-syndrome#synonyms
2)https://rarediseases.org/rare-diseases/kbg-syndrome/
3)https://rarediseases.info.nih.gov/diseases/82/kbg-syndrome
4) http://www.kbgfoundation.com/downloadable-graphics.html
NOT: KBG sendromluların 4 numaralı belirttiğim bir websiteleri mevcut. Hastalıkla ilgili bir posterleri var. Bunu Türkçeleştirmeyi ayrıca öneriyorum. İlgili kuruluşla iletişime geçip ‘’Bu posteri Türkçeleştirip kullanabilir miyiz’’ şeklinde sorulmalı. İlgili posteri aşağıya bırakıyorum. Mailde de ilgili posterin PDF halini göndereceğim, PDF çözünürlüğü yüksek.
