Genel Bilgi
Ürofasiyal sendrom ilk olarak 1960’larda ürolojik cerrah ve Kolombiya, Güney Amerika’dan araştırmacı Dr. Bernardo Ochoa tarafından tanımlanmıştır. Bozukluk Ochoa sendromu olarak da bilinir ve ilk başta bölgeye özgü olan yerel bir bozukluk olduğuna inanılıyordu. Ürofasiyal sendrom o zamandan beri dünya çapında ülkelerde ve etnik gruplarda tanımlanmıştır.
Ek anormallikler, böbreklerin ve pelvisin (piyelonefrit) iltihaplanması, idrarı böbrekten mesaneye (vezikoüreteral reflü) taşıyan tüplere geri akışını ve kabızlığı içerebilir. Hemen tanı ve tedavi, ciddi, geri dönüşü olmayan mesane ve böbrek hasarını azaltabilir veya potansiyel olarak önleyebilir. Akıl etkilenmez. Ürofasiyal sendrom, hastalığın bozulması veya değişikliklerinden (mutasyonlar) kaynaklanabilir.HPSE2 geni veya LRIG2 geni ve otozomal resesif bir şekilde kalıtsaldır.
Üriner anormallik, doğumda ortaya çıkabilecek idrar yollarının obstrüktif bir hastalığıdır (konjenital obstrüktif üropati). Bu üropati, mesane ve omurilik arasındaki sinir sinyallerinin başarısız olması nedeniyle mesanenin eksik boşalmasına (işeme) neden olur (nörojenik veya nöropatik mesane). Mesanenin en alt noktasında, idrarın çıkarıldığı tübüler yapı olan üretraya açılan dairesel bir kas lifi bandı (üretral sfinkter) vardır. Mesane idrarla dolduğunda, normalde omuriliğe sinyaller gönderilir. Daha sonra üretral sfinkterin gevşemesine neden olan sinir sinyalleri geri gönderilir. Ve mesane büzülür ve idrar yoluyla idrar gönderir. Bununla birlikte, etkilenen bireylerde, bilinmeyen nedenlerle bu tür sinir sinyallerinin başarısız olması sözkonusudur.
Görsel 1, kaynak: https://media.springernature.com/original/springer-static/image/chp%3A10.1007%2F978-1-4614-6430-3_241-2/MediaObjects/271938_0_En_241-2_Fig1_HTML.jpg
{kind=link}
Genetik Değişiklikler /Etken Faktörler
Ürofasiyal sendrom, iki farklı genden, HPSE2 geni veya LRIG2 geni mutasyonlarından kaynaklanır. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan proteinler oluşturmak için talimatlar sağlar. Bir genin mutasyonu meydana geldiğinde, protein ürünü hatalı, verimsiz veya olmayabilir. Belirli proteinin işlevlerine bağlı olarak, bu vücudun birçok organ sistemini etkileyebilir.
Ürofasiyal sendromlu bazı bireylerde, bilinen iki hastalık geninin hiçbirinde mutasyon yoktur, bu da henüz tanımlanmamış ek genlerin de bazı durumlarda bozukluğa neden olabileceğini düşündürmektedir.
Ürofasiyal sendrom otozomal resesif paternde kalıtsaldır. Resesif genetik bozukluklar, bir birey, durum için anormal genin iki kopyasını, her bir ebeveynden bir tane miras aldığında ortaya çıkar. Bir kişi hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için bir taşıyıcı olacaktır, ancak semptom göstermeyecektir. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Her hamilelikte ebeveyn gibi taşıyıcı bir çocuk sahibi olma riski% 50’dir. Bir çocuğun her iki ebeveynden de normal gen alma ve bu özellik için genetik olarak normal olma şansı% 25’tir. Risk erkekler ve kadınlar için aynıdır.
Belirti ve Semptomlar
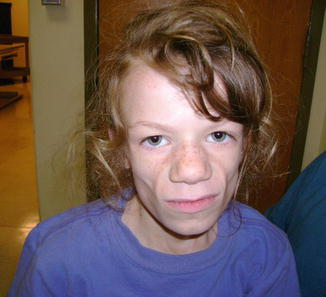
“Ürofasiyal” teriminin belirttiği gibi, bozukluk idrar ve yüz problemleri ile karakterizedir. Etkilenen bebeklerde ortaya çıkabilen ilk bulgu, alışılmadık bir “tersine çevrilmiş” yüz ifadesidir. Etkilenen bebekler gülmeye veya gülümsemeye çalıştığında, yüz kasları “ters çevirir”, böylece ekşitmeden veya ağlıyor gibi görünürler. Ürofasiyal sendromun semptomları ve şiddeti, aynı ailenin üyeleri arasında bile büyük ölçüde değişebilir.
Nörojenik mesane, normal olarak idrarı böbreklerden mesaneye (üreterler) getiren tüplere idrarın geri akışına (reflü) yol açabilir; üreterlerde (hidroüretre) ve böbreklerde (hidronefroz) anormal bir şişlik (distansiyon) ve idrar birikmesi. Etkilenen kişilerde hidroüreter ve hidronefroz hafif ila şiddetli arasında değişebilir. Bu tür anormalliklerin erken bir belirtisi, uyku sırasında gündüz ve / veya gece idrar tutamamayı (günlük ve gece enürezisi) içerebilir.
İdrar yollarının obstrüktif anormallikleri, idrar yolu hasarına ve tekrarlayan idrar yolu enfeksiyonlarına neden olabilir. Bazı durumlarda, bu tür enfeksiyonlar hiçbir belirtiye (asemptomatik) neden olabilir. Bununla birlikte, diğer durumlarda, sık idrara çıkma dürtüsü, aşırı miktarda idrarın (poliüri) geçişi, aşırı susuzluk (polidipsi), idrar geçerken yanma hissi, ağrılı veya zor idrara çıkma (ağrılı veya zor idrara çıkma) gibi çeşitli belirtiler ortaya çıkabilir (dizüri), mesane kontrolünün kaybı (inkontinans), genel bir hastalık hissi (halsizlik), ateş, alt karın ve / veya bellerde ağrı ve / veya şiddetli enfeksiyonlarda kan varlığı (hematüri) ve / veya idrarda irin. İdrar yolu enfeksiyonu, kan dolaşımına yayılabilir, bu da ürosepsis olarak bilinen ciddi bir komplikasyondur. Uygun tedavi olmadan, tekrarlayan idrar yolu enfeksiyonları ve böbrek yolunda kronik hasar nihayetinde kronik böbrek yetmezliğine yol açabilir. Böbrek yetmezliği, böbrekler atık ürünleri idrar yoluyla atma, vücuttaki tuz ve su dengesini düzenleme ve diğer hayati işlevlerini yerine getirme yeteneklerini kaybettiğinde ortaya çıkar. Sonunda böbrek yetmezliği gelişen etkilenen bireylerin tam oranı bilinmemektedir, ancak erken tedavi bu sonucun olasılığını azaltabilir. Bozukluğu olan bazı kişilerde böbrek hasarı nedeniyle yüksek tansiyon (hipertansiyon) olabilir. Bu ayrıca daha fazla böbrek hasarını önlemek için aktif tedavi gerektirir. vücuttaki tuz ve su dengesini düzenlemek ve diğer hayati işlevlerini yerine getirmek. Sonunda böbrek yetmezliği gelişen etkilenen bireylerin tam oranı bilinmemektedir, ancak erken tedavi bu sonucun olasılığını azaltabilir. Bozukluğu olan bazı kişilerde böbrek hasarı nedeniyle yüksek tansiyon (hipertansiyon) olabilir. Bu ayrıca daha fazla böbrek hasarını önlemek için aktif tedavi gerektirir. vücuttaki tuz ve su dengesini düzenlemek ve diğer hayati işlevlerini yerine getirmek. Sonunda böbrek yetmezliği gelişen etkilenen bireylerin tam oranı bilinmemektedir, ancak erken tedavi bu sonucun olasılığını azaltabilir. Bozukluğu olan bazı kişilerde böbrek hasarı nedeniyle yüksek tansiyon (hipertansiyon) olabilir. Bu ayrıca daha fazla böbrek hasarını önlemek için aktif tedavi gerektirir.
Etkilenen bireylerin yaklaşık üçte ikisi dışkının seyrek veya eksik geçişi yaşayabilir (kabızlık). Bazı çocuklar bağırsak hareketlerinin başarısız olmasının dışkının kolonda veya rektumda birikmesine neden olan bir durum olan şifrelemeyi geliştirebilir.
Etkilenen bazı kişiler, gece lagoftalmi olarak bilinen bir durum olan uyku sırasında göz kapaklarını tamamen kapatamazlar. Bu, uyanma üzerine kuru göz hissine, hafif veya şiddetli bir duyuma neden olabilir. Lagoftalmi, kornea iltihabı (keratit), çizik kornea (kornea aşınması), enfeksiyon ve kötü uyku gibi çeşitli komplikasyonlara neden olabilir.

Görsel 2, kaynak: http://www.scielo.br/img/revistas/eins/v13n2//1679-4508-eins-1679-45082015RC2990-gf02.jpg
{kind=link}
Genetik Görülme Sıklığı
Ürofasiyal sendrom her iki cinsiyeti de eşit olarak etkiler. Tıbbi literatürde 150’den fazla vaka bildirilmiştir. Literatüre göre, ailelerin çoğunluğu Kolombiya’dan gelmektedir, ancak etkilenen aileler ABD, Birleşik Krallık, Kuveyt, Danimarka ve İspanya’da da rapor edilmiştir.
Ürofasiyal sendrom gibi son derece nadir görülen bozukluklar genellikle tanınmadığından, bu bozukluklar yetersiz teşhis edilir, bu da genel popülasyondaki bu tür bozuklukların gerçek sıklığını belirlemeyi zorlaştırır.
Bazı araştırmacılar mesane, hidroüretre, hidronefroz ve ilişkili semptomlarda anormallikler yaşayan birçok bebek ve çocuğun ürofasiyal sendrom olabileceğinden şüphelenmektedir. Bununla birlikte, son araştırmalar, karakteristik yüz bulguları olmayan, fonksiyonel olmayan mesanesi olan etkilenen bireylerin, ürofasiyal sendrom kategorisinde olmayabilir, çünkü genetik kalıpları tipik olarak farklıdır.
Kalıtım Paterni / Deseni
Ochoa sendromu da denilen Urofacial syndrome, idrar sorunları ve olağandışı yüz ifadeleri ile karakterize bir hastalıktır.
Bu durum otozomal resesif paternde kalıtsaldır, yani her hücredeki genin her iki kopyasının mutasyonları vardır. Otozomal resesif koşulu olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Ochoa sendromuyla ilişkili idrar problemleri tipik olarak erken çocukluk veya ergenlik döneminde belirginleşir. Bu bozukluğu olan insanlar, idrar akışını (inkontinans) kontrol etmekte zorluk çekebilir ve bu da yatak ıslatmasına neden olabilir. Ochoa sendromlu bireyler mesaneyi tamamen boşaltamayabilir, genellikle idrarın normalde her böbrekten mesaneye ( üreterler) taşınan kanallara geri döndüğü bir durum olan vezikoüreteral reflü ile sonuçlanabilir.). İdrar da böbreklerde birikebilir (hidronefroz). Vezikoüreteral reflü (Görsel 3) ve hidronefroz, idrar yolu ve böbrek iltihabı (piyelonefrit) sık enfeksiyonlarına yol açabilir ve sonunda böbrek yetmezliğine neden olabilecek hasara neden olabilir.
Ochoa sendromlu bazı kişilerde HPSE2 geninde mutasyon yoktur . Bu kişilerde, bozukluğun nedeni bilinmemektedir.
Urofacial ochoa syndrome hastalığında prevelans bilinmemektedir.
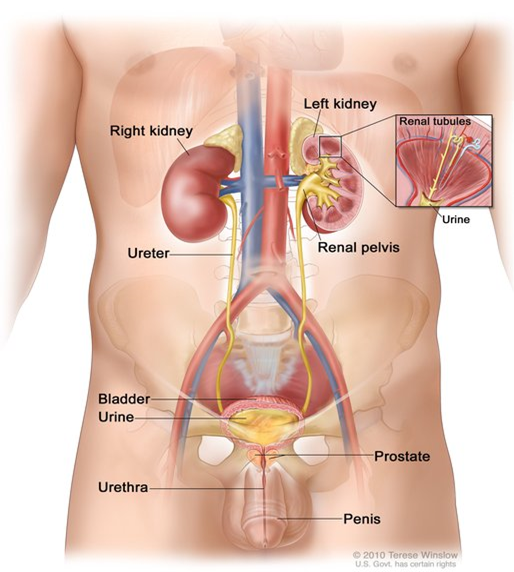
Görsel 3: https://ghr.nlm.nih.gov/art/large/male-urinary-system.jpeg
{kind=link}
Erkek üriner sistem: Böbrekleri, üreterleri, mesaneyi ve üretrayı gösteren erkek üriner sisteminin anatomisi. İdrar böbrek tübüllerinde yapılır ve her böbreğin böbrek pelvisinde toplanır. İdrar böbrekten üreterler yoluyla mesaneye akar. İdrar, vücudu üretradan terk edene kadar mesanede saklanır.
TEŞHİS YÖNTEMLERİ ve TEDAVİLERİ
Teşhis
Ochoa sendromlu bireyler, genellikle yüz ifadesinin ters çevrilmesi olarak tanımlanan, gülümsemeye veya gülmeye çalıştıklarında, kaşlarını çatlak karakteristik bir yüz buruşturma sergilerler. Bu özellik idrar yolu semptomlarından daha erken görünse de, belki bir bebek gülümsemeye başlar başlamaz, genellikle tıbbi müdahaleye getirilmez.
Ürofasiyal sendrom için resmi tanı kriterleri belirlenmemiştir. Tanı, karakteristik bulguların tanımlanması, ayrıntılı bir hasta ve aile öyküsü, kapsamlı bir klinik değerlendirme ve çeşitli özel testlere dayanarak şüphelenilir. Doktorlar, özellikle çocuk doktorları, nefrologlar ve ürologlar, üriner işeme anormalliklerinin ve bu bozukluğu karakterize eden benzersiz yüz ifadesinin ilişkisinin farkında olmalıdır.
Doğumda, etkilenen bebeklerde görünen ilk özellik “ters çevrilmiş” yüz
ifadesidir. Bu özelliğin güçlü bir ilişkisi ve ürolojik anormallikler
nedeniyle, ters yüz ifadelerinin varlığı idrar yolunun hızlı ve kapsamlı bir
incelemesine yol açmalıdır. Bu değerlendirme ürofasiyal sendromun erken
teşhisine ve spesifik ilişkili özelliklerin (örn. İdrar yolu enfeksiyonu) erken
teşhis edilmesine ve uygun önleyici adımların ve / veya hızlı tedavinin
sağlanmasında önemli bir rol oynayabilir.
Bu tür ürolojik değerlendirme sırasında, idrar yolu içindeki organların
yapısını incelemek ve işlevini değerlendirmek için özel görüntüleme teknikleri
kullanılabilir (örn. Böbreklerin, mesanenin, üreterlerin farklı
kısımları). Bu tür testler, özel bir kontrast ortamının enjekte edildiği
ve x-ışınlarının alındığı intravenöz piyelografiyi (IVP) ve idrar yolundan
sıvıların hareketini ve akışını inceleyen ürodinamik değerlendirmeyi
içerebilir.
Nöropatik mesaneye ve obstrüktif anormalliklere ek olarak, bu tür özel görüntüleme çalışmaları da idrar yolunun yapısal anormalliklerini ortaya çıkarabilir. Mesanenin anormal, ankraj bantları ve bağ dokusu kordonları (trabekülasyon) olabilir ve mesanenin kasından küçük keseye benzer çıkıntılara (herniler) ve aşırı kese benzeri çıkıntılara (herniler) neden olabilir. duvar (divertikül). Ek olarak, mesanenin dış kas tabakası (detrusor kası) alışılmadık derecede artmış refleks reaksiyonları (hiperrefleksi) ve anormal, uzun süreli kasılmalar (hipertonik kontraktürler) gösterebilir; mesanenin boynu anormal şekilde büyüyebilir (hipertrofik); ve / veya üretra düzensiz spazmlara (spastik üretra) sahip olabilir ve anormal bir çapa (kalibre) sahip olabilir.
İdrar yolu enfeksiyonları, klinik değerlendirme, mikroskobik inceleme ve idrar örneklerinin bakteriyolojik kültürü ile teşhis edilebilir.
Moleküler genetik test, ürofasiyal sendrom tanısını doğrulayabilir. Moleküler genetik test, hastalığa spesifik genlerde (yani HPSE2 veya LRIG2 ) mutasyona neden olduğu bilinen mutasyonları tespit edebilir , ancak sadece uzmanlaşmış laboratuvarlarda bir teşhis servisi olarak mevcuttur.
Doğum öncesi tanı ürofasiyal sendrom öyküsü olan ve hastalığa neden olan gen mutasyonunun bilindiği ailelerde mümkündür.
Tedavi
Ürofasiyal sendromun tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, bir uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, idrar yolu bozukluklarının teşhisi ve tedavisi konusunda uzmanlaşmış doktorlar (ürologlar), böbrek bozuklukları (nefrologlar) konusunda uzmanlaşmış doktorlar, cerrahlar, diyetisyenler ve / veya diğer sağlık uzmanları, etkilenen bir çocuğun sistematik ve kapsamlı bir şekilde planlanması gerekebilir tedavisi. Genetik danışmanlık etkilenen bireyler ve aileler için faydalıdır.
Ürofasiyal sendromun hemen tanınması ve erken tedavisi, ciddi vakalarda ortaya çıkabilecek potansiyel olarak ciddi, geri dönüşümsüz mesane ve böbrek hasarının azaltılması veya önlenmesinde kritik öneme sahiptir.
İdrar yolu enfeksiyonlarının tedavisi genellikle bakteriyel enfeksiyonların ve ağrı kesicilerin (örn. Analjezikler) tedavisi ve önlenmesi için antibiyotik içerir. Bazı insanlarda, idrar yolu tıkanıklığını ortadan kaldırmak ve idrar yolunun belirli kısımlarını (örn. Üreterler, üreterovesiküler kavşak) yeniden yapılandırmak için ameliyat yapılabilir. Yapılan cerrahi prosedürler anatomik anormalliklerin şiddetine ve ilişkili semptomlarına bağlıdır.
Ürofasiyal sendromlu bireyleri tedavi etmek için spesifik ilaçlar, anti-kolinerjik ve alfa-1-adrenerjik blokerler kullanılmıştır. Kabızlık, genel popülasyonda olduğu gibi standart kılavuzlarla tedavi edilmelidir.
Mesanenin tam boşaltımı sağlanamadığı durumlarda, Aralıklı kateterizasyon (T.A.K. temiz aralıklı kateterizasyon) gerekebilir. Üst üriner sistem bozulmasını ve böbrek yetmezliğini önlemek için erken tanı ve tedavi gereklidir.
Kronik böbrek yetmezliği yaşayan etkilenen çocuklarda, tedavi seçenekleri, fazla atık ürünleri düzenli olarak kandan (diyaliz) uzaklaştıran belirli prosedürleri içerebilir. Bu prosedürler, bir makine yoluyla kanı süzerek (hemodiyaliz) ve / veya vücudun karnında doğal bir filtreleme membranı (periton diyalizi) kullanarak atıkların giderilmesini içerebilir. Bazı ciddi böbrek yetmezliği vakalarında böbrek nakli düşünülebilir.
Etkilenen çocukların potansiyellerine ulaşmasını sağlamak için erken müdahale önemlidir. Yararlı olabilecek özel hizmetler, özel sosyal desteği ve diğer tıbbi ve / veya sosyal hizmetleri içerebilir.
Hastalıkla İlişkili Genler
Aşağıdaki bozuklukların belirtileri ürofasiyal sendromdakilere benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Hinman-Allen sendromu veya nörojenik olmayan nörojenik mesane olarak da bilinen Hinman sendromu, nörolojik bir eksiklik olmadığı için nöropsikolojik kökenli olduğuna inanılan nadir bir işeme bozukluğudur. Etkilenen bireyler, ortaya çıkmayan yüz ifadesinde anormallikler dışında, ürofasiyal sendromlu bireylerde görülenlere oldukça benzer klinik özellikler sergiler.
Ürofasiyal sendroma ek olarak, ek konjenital bozukluklar nörojenik mesane, hidroüretre ve / veya hidronefroz gibi idrar yolu anormallikleri; etkilenen erkeklerde kriptorşidizm gibi genital sistem malformasyonları; yüz anormallikleri; ve / veya ürofasiyal sendromla potansiyel olarak ilişkili olanlara benzer diğer fiziksel anormallikler.
Hastalığın Diğer İsimleri
- Ochoa sendromu
- tuhaf yüz ifadesi ile hidronefroz
- ters gülümseme ve gizli nöropatik mesane
- üriner anormallikleri olan kısmi fasiyal palsi
- UFS
- ürofasiyal Ochoa sendromu
Kaynaklar