Genel Bilgi
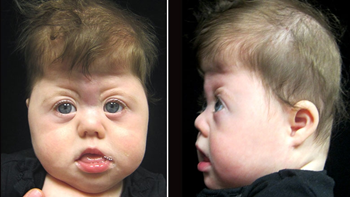
Saethre-Chotzen sendromu, bazı kafatası kemiklerinin (krainosinostoz) erken füzyonu ile tanımlanan genetik bir durumdur. Bu erken füzyon kafatasının normal şekilde büyümesini önler ve kafa ile yüzün şeklini ve simetrisini etkiler. Genellikle koni şeklinde bir kafa, asimetrik yüz, saç hizasının aşağıda olması ve sarkık göz kapakları (ptosis) şeklinde tanımlanan bir sendromik kraniyosinostoz şeklidir.
Görsel Kaynağı: https://www.chop.edu/conditions-diseases/saethre-chotzen-syndrome
Genetik Değişiklikler/Etken Faktörler
SCS’u hücre soyunun belirlenmesiden ve farklılaşmasından sorumlu temel bHLH kopyalama faktörünü kodlamakla görevli TWIST1 (7p21) geninin nokta mutasyonlara uğraması veya silinmesi nedeniyle gerçekleşir. Bu gendeki mutasyonlara erken kafatası birleşmesine eden olur. Gen silmeleri genellikle önemli nörokognitif gecikmelerle ilişkili olan daha ciddi fenotiplere neden olur. FGFR3, FGFR2 and TCF12’deki mutasyonların, fenotip olarak SCS ile örtüşen sinostoz koşullarına neden olduğu rapor edilmiştir.
Belirti ve Semptomlar
SCS’ nun belirti ve semptomları aynı aileden etkilenen bireyler arasında bile geniç ölçüde çeşitlilik gösterir.Bu durum, ellerde ve ayaklarda hafif anormalliklere neden olabilir örneğin her bir eldeki ikinci ve üçüncü parmakların birleşmesi ve geniş veya kopyalanmış büyük ayak parmağı gibi. Gecikmiş gelişme ve öğrenme zorluğu bildirilmiştir, ancak bu duruma sahip çoğu kişi normal zekaya sahiptir. Çok yaygın olmayan belirti ve semptomlarından bazılarıda kısa boyluluk, omurga kemiklerindeki anormallikler, işitme kaybı ve kalp hasarıdır.
Genetik Görülme Sıklığı
Saethre-Chotzen sendromu tahmini 25.000 ila 50.000 kişide bir görülme sıklığına sahiptir.
Kalıtım Paterni/Deseni
Bu durum otozomal dominant şekilde kalıtılır, bu durumda da her hücredeki değiştirilmiş genin bir kopyası bozukluğa neden olur. Bazı durumlarda, etkilenen kişi etkilenen bir ebeveynin mutasyonunu devralır. Diğer durumlar, gendeki yeni mutasyonlardan kaynaklanabilir. Bu vakalar ailelerinde herhangi bir bozukluk olmayanlarda görülür.
Bazı insanlar TWIST1 mutasyonlı oldukları halde SCS’unun belirgin bir özelliğine sahip değillerdir. Bu insanlar hala bu gen mutasyonunun çocuklarına geçme ve diğer belirtileri ve semptomları gösterme riski altındadırlar.
Teşhis Yöntemleri ve Tedaviler
SCS tedavisi, kraniyofasiyal bir ekip tarafından genç erişkinliğe kadar takip gerektirir. Genel olarak, hastalar intrakraniyal hacmi arttırmak ve anormal bir kafa şeklini eski haline getirmek için yaşamın ilk yılından bir kraniyoplasti (kafatasının bir kusurunun veya deformasyonunun cerrahi onarımı) yaptırmalıdır.
Çocukluk çağında, solunum yolu tıkanıklığı ve maloklüzyon tedavisi için orta yüz cerrahisi gerekli olabilir. Yarık damaktakilerde, diğer malformasyonlar ve gerekli olan konuşma terapisi bağlamında cerrahi kapatma yapılabilir. Yüz büyümesinin, işitme kaybının ve psikomotor gelişimin rutin değerlendirmelerine ek olarak, şaşılık, ambliyopi veya kronik papilödem (artmış ICP’yi gösterir) izlemek için düzenli oftalmolojik muayenelere ihtiyaç vardır. Gelişimsel gecikme olan çocuklara erken müdahale programları önerilmelidir.
Hastalıkla İlişkili Genler
TWIST1
FGFR2
Hastalığın Diğer İsimleri
- Acrocephalosyndactyly III
- Acrocephalosyndactyly, type III
- Acrocephaly, Skull Asymmetry, and Mild Syndactyly
- ACS III
- ACS3
- Chotzen senndromu
- Hipertelorizmli disostoz kraniyofasiyal
- SCS
Kaynaklar
Scott P. Bartlett, MD, Jesse A. Taylor, MD (https://www.chop.edu/conditions-diseases/saethre-chotzen-syndrome)