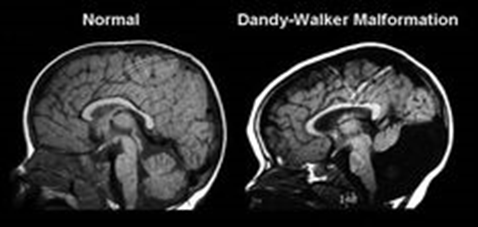
Tanım
Ritscher-Schinzel sendromu olarak da bilinen 3C sendromu, kraniyofasiyal anormallikler, konjenital kalp kusurları ve serebellar beyin malformasyonları görülen gelişimsel bir malformasyon sendromudur.
Klinik Tanım
3C sendromu, azalan sıklıkla, alçak konumlu kulaklar, hipertelorizm, aşağı eğik palpebral fissürler, basık burun köprüsü, belirgin oksiput, belirgin alın, mikroginati, oküler kolobom, yarık damak gibi belirgin kraniyofasiyal özellikler ile taşıyan konjenital bir hastalıktır. Ek kraniyofasiyal özellikler, nevus flammeus’u (alın bölgesini) kapsar. arka saç çizgisini, seyrek kafa derisi saçları, kaşları ve kirpikleri, çıkıntılı dille açık ağız ve kısa boyunu içerir. Olguların% 80’inde serebellar anomaliler vardır ve bunlar arasında öncelikle serebellar vermis hipoplazisi ve sisterna magmanın genişlemesi yer alır. Etkilenen bireylerde hantal motor, zihinsel engel ve konuşma gecikmesi vardır. Kardiyovasküler anomaliler arasında atriyal ve ventriküler septal kusurlar, patent arteriyel kanal, Fallot tetralojisi, çift çıkışlı sağ ventrikül, hipoplastik sol kalp sendromu , aortik veya pulmoner stenoz ve diğer kapak anomalileri bulunur. Birçok hastada doğum sonrası kısa boyluluk ve 2 hastada da büyüme hormonu eksikliği rapor edilmiştir. Hastaların >%10’unda görülen ek anomaliler arasında beslenme güçlükleri, tek göbek arteri, tek enine kırışıklı küçük eller, kamptodaktili, ekinovarus bozukluğu, hidronefroz, yüzeysel skrotum, inmemiş testis, kriptorşidizm, mikropenis, hipospadias, tırnak hipoplazisi, işitme kaybı, bağırsakta malrotasyon. İskelet kusurları kaburga ve vertebral anomaliler ile ortaya çıkabilir. Nadiren gözlenen özellikler arasında oküler (konjenital glakom, göz kapağı pitozu ile optik sinir atrofisi, heteokromatik iris, posterior embriyotokson), böbrek (multisistik displastik böbrek, at nalı böbrekler, tek taraflı böbrek agenezi) ve anal (anal atrezi, anterior yerleşimli anüs malformasyonları) anomalileri vardır. Renal hipoplazi, meme başı hipoplazisi, penil hipoplazi, tek taraflı adrenal aplazi,immün yetmezlik rapor edilmiştir.
Epidemiyolojisi
Bugüne kadar <50 vaka tanımlanmıştır. Sendrom panetnik görünüyor.
Kalıtım Modeli
Dandy-Walker malformasyonu vakalarının çoğu sporadiktir, bu da ailelerinde kusurlu olma durumunun olmadığı kişilerde ortaya çıktığı anlamına gelir. Vakaların küçük bir yüzdesi ailelerde de görülüyor ancak, Dandy-Walker malformasyonunun açık bir miras modeli yoktur. Birden fazla genetik ve çevresel faktör muhtemelen bu bozukluğun gelişme riskini belirlemede rol oynamaktadır. Dandy-Walker malformasyonuna sahip kişilerin birinci dereceden akrabalarında (kardeşler ve çocuklar gibi) genel popülasyondaki insanlara kıyasla daha yüksek oranda görülür.
Yönetim ve Tedavi
Yönetim esas olarak eğitim programları, fiziksel, mesleki ve konuşma terapisini içeren semptomatik ve multidisipliner yaklaşımlardır. Hipotoni ve gelişmeye yönelikmotor gecikmesini azaltmayı güçlendirmek için yapılan kardiyak malformasyonlar özel bakım gerektirir, genellikle ameliyattır.
Prognoz
Prognoz kardiyovasküler malformasyon ile belirlenir. Motor gecikmesi yaygındır ve serebellar anomalilere sekonder hipotoni ile ilişkilidir.
Hastalıkla İlgili Genler
3C/Ritscher-Schinzel sendromu doğuştan kraniyo-serebello-kardiyak displazi’ye sebep olan, 11.kromozomunda görülen mutasyona sebep olan CCDC22 ve WASHC5 genleriyle alakalıdır.
Hastalığın Diğer İsimleri
- Dandy-Walker kompleksi
- Dandy-Walker kisti
- Dandy-Walker şekil bozukluğu
- Dandy-Walker sendromu
- DWM
- DWS
- hidrosefali, iç, Dandy-Walker tipi
- hidrosefali, iletişimsiz, Dandy-Walker tipi
- Luschka-Magendie foramina atrezi
- Kranyo-serebello-kardiyak displazi
- 3C sendromu
- Ritscher Schinzel sendromu
- Ritscher-Schinzel kranyo-serebello-kardiyak sendrom
Kaynaklar