Fraiser sendromu, psödohermafroditizm ve progresif glomerülopati ile tanımlanan nadir bir hastalıktır.Hastalar normal kadın dış genital organları, çizgi gonadları ve XY karyotipi ile başvurur ve sıklıkla gonadoblastom gelişir. Glomerüler semptomlar, ergenlik veya erken yetişkinlikte son dönem böbrek yetmezliğine ilerleyen spesifik olmayan fokal ve segmental glomerüler skleroz ile karakterize çocukluk çağı proteinüri ve nefrotik sendromdan oluşur.
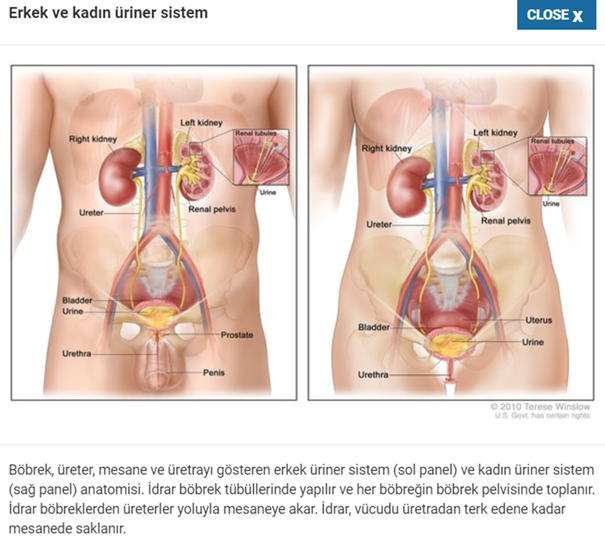
Frasier sendromu böbrekleri ve genital organları etkileyen bir durumdur. Etkilenen bireyler, bazı glomerüllerde skar dokusunun oluştuğu, böbreklerdeki kanı atıktan süzen küçük kan damarları olan skar dokusunun oluştuğu fokal segmental glomerüloskleroz adı verilen bir duruma sahiptir.Frasier sendromlu kişilerde ,bu durum genellikle ergenlik döneminde böbrek yetmezliğine yol açar.
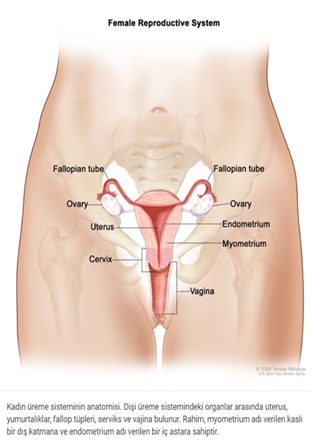
Frasier sendromlu erkeklerde tipik erkek kromozom paterni olmasına rağmen ( 46, XY), dış genital organların açıkça erkek veya açıkça kadın (belirsiz genital) veya genital organların tamamen dişi göründüğü gonadal disgenezisi vardır. İç üreme organları (gonadlar) tipik olarak gelişmemiş ve çizgi gonadları olarak adlandırılmıştır. Bu anormal gonadlar işlevsizdir ve genellikle kanserli hale gelir, bu nedenle genellikle ameliyatın erken dönemlerinde çıkarılırlar.
Etkilenen dişilerin genellikle normal cinsel organları ve gonadları vardır ve durumun sadece böbrek özelliklerine sahip. Durumun tüm özelliklerine sahip olmadıkları için, kadınlara genellikle izole nefrotik sendrom tanısı verilir.

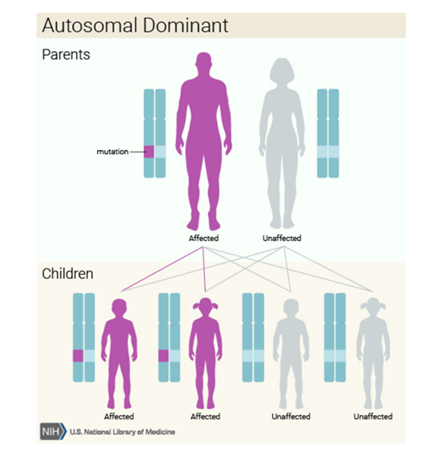
Kalıtım Kalıbı
Bu durum otozomal dominant bir paternde kalıtsaldıryani her hücredeki değiştirilmiş genin bir kopyası bozukluğa neden olmak için yeterlidir.
Klinik Tanım
Moorthy ve diğ. (1987) , Denys-Drash sendromu vakaları olarak bildirilen bazı hastaların aslında Fraiser sendromu adını önerdikleri farklı bir bozukluğa sahip olduklarını ileri sürmüşlerdir. Moorthy ve diğ. (1987) , daha önce bildirilen 6 çizgi gonad, psödohermafroditizm ve böbrek yetmezliği olan hastayı tartışmıştır. Bazı hastalarda tanı, primer amenore değerlendirmesi sırasında başarılı bir böbrek nakli sonrasında konmuştur. 6 hastanın 5’inde çizgi gonaddan kaynaklanan gonadoblastom saptandı.
Fraiser sendromu ile ilişkili 3 hasta tanımlamışt. Her üçü de 2-6 yaş arasında kalıcı proteinüri ile başvurdu ve daha sonra 9-35 yaşları arasında son dönem böbrek yetmezliğine ilerleyen nefrotik sendrom gelişti. Böbrek yetmezliğinin başlamasından önce yapılan böbrek biyopsilerinde 1 hastada minimal nonspesifik glomerüler değişiklikler, diğer 2 hastada fokal ve segmental glomerüler skleroz saptandı. Her üç hastaya da nefrotik sendrom nüksü olmadan başarılı böbrek nakli yapıldı. Normal kadın fenotipi olan bu 3 kadında primer amenore değerlendirmesi 46, XY gonadal disgenezis tanısına yol açmıştır. Üç hastadan birinde 19 yaşında teşhis edilen gonadoblastom gelişti; cerrahi tedaviden sonra nüks gözlenmedi. Diğer 2 hastaya bilateral cerrahi gonadektomi yapıldı.
Melo ve diğ. (2002) , Denys-Drash sendromunun dış genital özelliğine sahip Fraiser sendromu tanısı alan bir hastayı bildirmiştir. Bu 2 sendromunun farklı hastalıklar olmadığını, ancak WT1 genindeki değişikliklerin neden olduğu bir spektrumun 2 ucunu temsil edebileceğini öne sürdüler.
Görülme Sıklığı
Frasier sendromunun nadir bir durum olduğu düşünülmektedir; bilimsel literatürde yaklaşık 50 vaka tanımlanmıştır.

Moleküler Genetik
WT1 geninin ekson 8 veya 9’a mutasyonlar Denys-Drash 10 ilgisiz hastalarda tespit edilmiş olduğundan sendromu ile Pelletier et al.(1991) , Fraiser sendromlu hastalarda WT1’in 1 ila 10 eksonlarını taradılar , ancak hiçbir mutasyon tespit etmediler. Tek iplikli konformasyon polimorfizm (SSCP) analizi ile mutasyon taraması yaptıkları için, nokta mutasyonlarının sadece tahmini %80’ini tespit eden bir yöntem olduğundan, genin başka yerlerinde mutasyon olasılığı tamamen dışlanmamıştır.
Berta ve diğ. (1992) , XY gonadal disgenezisi ve kronik böbrek yetmezliği olan 2 kız çocuğunda Y kromozomunun cinsiyet belirleyici bölgesinde veya SRY genindeki mutasyonlarda ( 480000 ) büyük bir delesyon bulamadılar .
3 hastada Fraiser sendromu, Barbaux ve diğ. (1997) , WT1 geninin intron 9’unun ( 607102.0018 ; 607102.0019 ) donör ekleme alanındaki mutasyonları, + KTS izoformu olarak bilinen bir kayıp ile tanımladılar . Normal olarak, intron 9’daki alternatif bir ekleme yeri, WT1 proteininin üçüncü ve dördüncü çinko parmakları arasına 3 amino asit (KTS) eklenmesine izin verir. 3 hastanın hepsinde erkek psödohermafroditizmi, nefrotik sendrom vardıson dönem böbrek yetmezliğine ilerleyen ve 46, XY gonadal disgenezi. Böbrek yetmezliğinin başlamasından önce yapılan böbrek biyopsilerinde 1 hastada minimal nonspesifik glomerüler değişiklikler, diğer 2 hastada fokal ve segmental glomerüler skleroz saptandı. Bir hastada 19 yaşında gonadoblastom teşhis edildi.
Barbaux ve diğ. (1997) , hastanın Denys-Drash sendromu olduğu ve WT1 geninin intron 5’inde ( 607102.0009 ) mutasyonun aslında Frasier sendromu olduğunu bildirmiştir .
Klamt ve diğ. (1998) , Fraiser sendromuna neden olan WT1 mutasyonları tarafından hiçbir mutant proteinin üretilmediğini göstermiştir . Bunun yerine, mutasyon, proteinin 2 ilave izoformunun, fazladan 3 amino aside (KTS) sahip olan ve olmayanlara değiştirilmiş bir oranla sonuçlanır. Denys-Drash sendromunda, tümör riski Fraiser sendromundan çok daha fazladır . Baskın negatif mutant aleli arızalıdır ve ikinci alelin kaybı (2 vuruşlu modele göre) tümör oluşumunda önemli bir adım olabilir. Aksine, Fraiser hastaların sadece daha kısa bir izoform üretebilen 1 normal WT1 ve 1 kopyası vardır. Bu nedenle alel kaybı, WT1’in + KTS izoformunu üretemeyen, ancak yine de büyük miktarlarda -KTS izoformuna sahip olan hücrelere yol açacaktır. Bu bakımdan, çıplak farelerde G401 Wilms tümör hücre dizisinin tümör oluşumunun + KTS ve -KTS izoformları ile aynı ölçüde bastırılabileceğini belirtmek ilginçtir. Gonadoblastoma sık olduğu Frasier hastalar.
Melo ve diğ. (2002) , bağlanma yeri kullanımında bir değişiklik öngören IVS9 + 4C-T mutasyonu ( 607102.0018 ) olan Fraiser sendromlu 19 yaşında bir erkek olduğunu bildirmiştir . Sıradışı bir fenotipi vardı. WT1 transkript analizi FS tanısını doğrulayan normal pozitif / negatif KTS izoform oranının tersine döndüğünü gösterdi. Yazarlar, bu hastanın Denys-Drash sendromun dış genital özelliğine sahip olduğu ve bu 2 sendromun ayrı hastalıklar olmadığını, ancak WT1 genindeki değişikliklerin neden olduğu bir spektrumun 2 ucunu temsil edebileceği sonucuna vardı .
Nedenler
WT1 genindeki mutasyonlar Frasier sendromuna neden olur . WT1 geninin spesifik bölgelerine bağlanması (bağlanma) ile, diğer genlerin aktivitesini düzenleyen bir protein yapmak için yönergeler DNA. Bu eyleme dayanarak, WT1 proteinine transkripsiyon faktörü denir. WT1 proteini böbreklerin ve gonadların gelişiminde rol oynar (kadınlarda yumurtalıklar ve erkeklerde testisler) doğumdan önce.
Frasier sendromuna neden olan WT1 gen mutasyonları, gen aktivitesini kontrol etme ve böbreklerin ve üreme organlarının gelişimini düzenleme yeteneğine sahip bir proteinin üretilmesine yol açarak Frasier sendromunun belirti ve semptomlarına neden olur.
Frasier sendromu, WT1 genindeki mutasyonların da neden olduğu Denys-Drash sendromu adı verilen başka bir duruma benzer özelliklere sahiptir . Bu iki durum genetik bir nedeni paylaştığı ve üst üste binen özelliklere sahip olduğu için, bazı araştırmacılar bunların iki farklı koşulun değil, bir spektrumun parçası olduğunu ileri sürmüşlerdir.
Normal İşlev
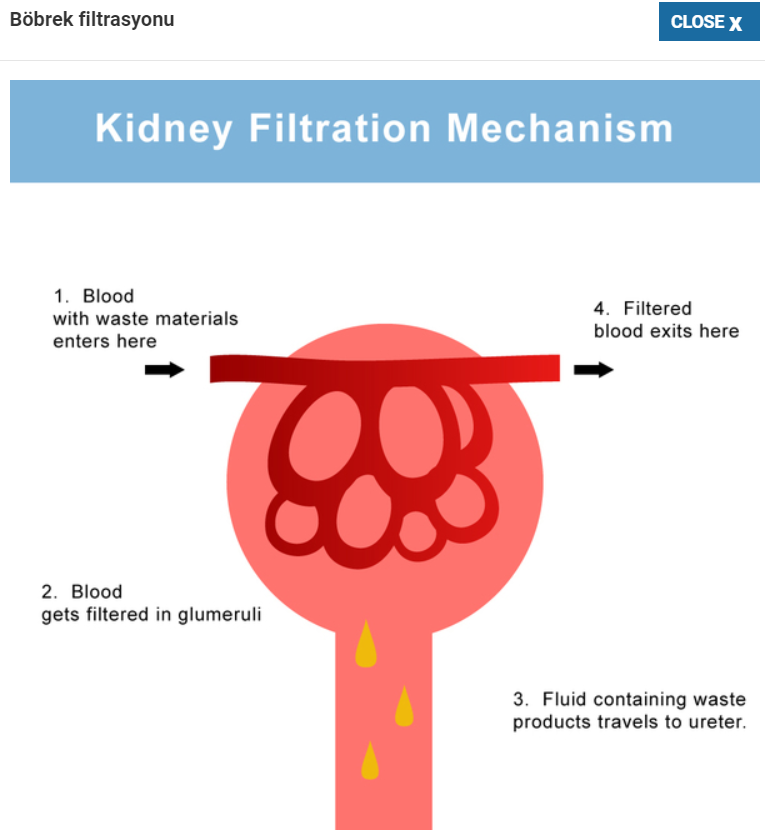
WT1 geni doğumdan önce (erkeklerde kadınlarda yumurtalıkların ve testislerin) böbrekler ve üreme organlarına gelişimi için gerekli olan bir proteini yapmak için talimatlar sağlar. Doğumdan sonra, WT1 protein aktivitesi glomerulus olarak bilinen ve böbrekler yoluyla kanı süzen bir yapı ile sınırlıdır. WT1 proteini, hücre büyümesinde, hücrelerin belirli işlevleri yerine getirmek için olgunlaştıkları (farklılaşma) ve hücrelerin kendi kendini yok etmelerinde (apoptoz) rol oynar. Bu işlevleri yerine getirmek için WT1 proteini, DNA’nın belirli bölgelerine bağlanarak (bağlanarak) diğer genlerin aktivitesini düzenler. Bu eyleme dayanarak, WT1 proteinine transkripsiyon faktörü denir.
Etolojisi
Frasier sendromu, WT1 geninde (11p13) intron 9’un (önceden IVS9 + 4; IVS9 + 5 olarak anılacaktır) 4-5 nükleotidlerini etkileyen spesifik patojenik varyantlarla ilişkilendirilmiştir. WT1, hem böbrek hem de gonadal gelişim için önemli olan düzenleyici transkripsiyon faktörü olarak işlev gören bir proteini kodlar.
Hastalık Belirtileri
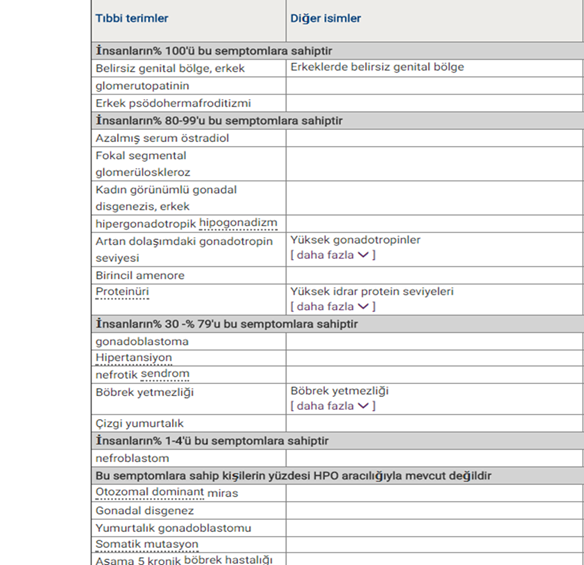
Bu tablo, bu hastalığı olan kişilerin sahip olabileceği belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye değişir. Aynı hastalığı olan insanlar listelenen tüm semptomlara sahip olmayabilir. Bu bilgiler İnsan Fenotip Ontolojis (HPO) adı verilen bir veritabanından gelir.. HPO, tıbbi kaynaklarda açıklanan semptomlar hakkında bilgi toplar. HPO düzenli olarak güncellenir. Bir belirti hakkında daha ayrıntılı bilgiye erişmek için HPO Kimliğini kullanın
Teşhis Yöntemleri
Histolojik analizde FSGS bulguları ile ilerleyen glomerulopatinin çocukluk başlangıcında tanıdan şüphelenilmektedir. Ergenlik gecikmesi veya primer amenore olan fenotipik dişiler nefropati belirtileri açısından dikkatle değerlendirilmelidir. Klinik bulgular WT1 ile ilişkili bozuklukların teşhisini önerdiğinde, sıcak noktanın 8-9 tek gen testieksonbitişik intronlar ile gerçekleştirilebilir. Karyotip testi, WT1 intron 9 patojenik varyantları olan tüm bireyler için önerilir.
Genetik veya nadir bir hastalık için teşhis koymak genellikle zor olabilir. Sağlık uzmanları, bir tanı koymak için genellikle bir kişinin tıbbi geçmişine, semptomlarına, fiziksel muayenesine ve laboratuvar test sonuçlarına bakar. Aşağıdaki kaynaklar, bu durumun teşhisi ve testi ile ilgili bilgi sağlar. Teşhis hakkında sorularınız varsa, bir sağlık uzmanıyla görüşmelisiniz.
Yöntem ve Tedavi
Yönetim çok disiplinlidir ve aşağıdakileri içermelidir: nefrolog kronik böbrek yetmezliğinin tedavisi için (başlangıçta nefroprotektif tıbbi tedavi ile ve daha sonra ESRD oluştuğunda böbrek replasman tedavileri veya transplantasyon ile), endokrinologlar ilişkili testis gelişimi bozukluğunun tedavisi için ve onkologlarve cerrahlar tümör oluşumunu önlemek için erken bir gonadektomi ihtiyacını değerlendireceklerdir. Böbrek nakli veya peritoneal yerleşim sırasında preemptif bilateral gonadektomidiyaliz sonda bir seçenek olabilir.
Prognoz
Yaşam beklentisi hakkında sınırlı bilgi vardır. Böbrek nakli sonrası nefrotik sendrom tekrarlamaz. 46, tam gonadal disgenezi olan XY bireyleri kısırdır.
Kaynaklar
Yaşam beklentisi hakkında sınırlı bilgi vardır. Böbrek nakli sonrası nefrotik sendrom tekrarlamaz. 46, tam gonadal disgenezi olan XY bireyleri kısırdır.