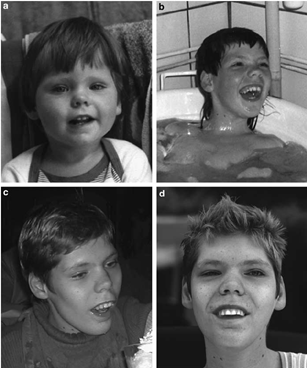
Genel Tanım
Allan – Herndon – Dudley sendromu (AHDS) ciddi derecede zihinsel gelişim, dizartri, atetoid hareketler, kas hipoplazisi ve spastik parapleji ile karakterize X’e bağlı bir durumdur. Sadece erkeklerde meydana gelen bu durum, gelişimi doğumdan önce bozar. Hastalık, etkilenen erkeklerde konuşmayı ve iletişim yeteneğini kısıtlamış olsa da, diğer insanlarla iletişimde eğleniyor gibi görünüyorlar.
Allan-Herndon-Dudley sendromlu çocukların çoğunda zayıf kas tonusu (hipotoni) ve birçok kasın az gelişmesi (kas hipoplazisi) durumu vardır. Yaşlandıkça, genellikle belirli eklemlerin hareketini kısıtlayan kontraktür denilen eklem bozuklukları geliştirir. Anormal kas sertliği (spastisite), kas zayıflığı ve kolların ve bacakların istemsiz hareketleri de hareketliliği sınırlar. Sonuç olarak, Allan-Herndon-Dudley sendromlu birçok insan bağımsız olarak yürüyemez ve yetişkinlikte tekerlekli sandalye kullanımı mevcuttur.
Klinik Tanım
Hastalık spastisiteye (kontraktürler, Babinski işareti ve klonus) ilerleyen ve genellikle yaşamın erken döneminde saptanabilen konjenital hipotoni (doğumda veya yaşamın ilk haftalarında / aylarında görülür) olarak kendini gösterir. Hiperrefleksi, daha sonra yaşamda ortaya çıkar. Etkilenen erkekler de bebeklik ve erken çocukluk döneminde, motor dönüm noktaları gecikmesi ve baş ve desteklemede zorluk olarak ortaya çıkan kas hipoplazisi ve genel kas zayıflığı ile kendini gösterir. Hastaların% 100’ünde hipotoni ve ciddi zihinsel eksiklik vardır. Şiddetli psikomotor gecikme en başından itibaren mevcuttur (motor ve dil dönüm noktalarının gecikmesi) ve özerkliğe asla ulaşılamaz. Yüz, zaman içinde gelişen ayırt edici özelliklere sahiptir: açık ağız, çadırlı üst dudak, pitoz(sarkma) , kulakların anormal katlanması, burun ve kulakların yumuşak dokusunun kalınlaşması ve kalkık kulak memeleri. Uzun, ince eğik ayaklar da tipiktir. Oküler belirtiler (yani döner nistagmus ve ayrık göz hareketleri) nadirdir. Nöbetler ve kilo alım problemleri bazı hastalarda bildirilmiştir. Hipotoni ve kas hipoplazisinin bir sonucu olarak düşünülen , pektus ekskavatum ve skolyoz bulunabilir.
Epidemeyoloji
Bugüne kadar literatürde 320 hastalıktan etkilenen bireye sahip en az 132 aile bildirilmiştir. Yaygınlık bilinmemekle birlikte, bir çalışma, etiyolojisi bilinmeyen zihinsel özürü olan erkeklerin% 1.4’ünde AHDS’yi tanımlamıştır. Sadece erkekler etkilenir.
Kalıtım Kalıbı
Bu durum X’e bağlı resesif, kalıtsaldır. Hastalığa neden olan mutasyona uğramış gen , iki cinsiyet kromozomundan biri olan X kromozomu üzerinde bulunuyorsa, durum X’e bağlı olarak kabul edilir. Sadece bir X kromozomu olan erkeklerde , her hücrede genin değiştirilmiş bir kopyasının bulunması, hastalığa neden olmak için yeterlidir. İki X kromozomu olan kadınlarda, bozukluğa neden olmak için genin her iki kopyasında da mutasyon bulunmalıdır. Erkekler X’e bağlı resesif bozukluklardan kadınlardan daha sık etkilenir. X’e bağlı kalıtımın bir özelliği, babaların X’e bağlı özellikleri, oğullarına geçirememesidir.
Teşhis Yöntemleri
Tanı, klinik bulgulara ve diğer tiroid hormonu serum düzeylerinin varlığına dayanır: erkeklerde anormal derecede yüksek 3,3 ‘, 5’-triiyodotironin (T3), düşük ila normal serbest tetraiyodotironin (T4) seviyeleri ve normal ila hafif yüksek tiroid uyarıcı hormon (TSH) seviyeleri vardır. SLC16A2 genindeki mutasyonları gösteren moleküler genetik test tanıyı doğrular.
Yönetim ve Tedavi
Şu anda, AHDS için herhangi bir tedavi mevcut değildir ve yönetim destekleyici önlemlerden oluşur. Fiziksel, mesleki ve konuşma terapisi faydalı olabilir. Distoni, antikolinerjikler, L-DOPA, karbamazepin veya lioresol gibi bazı ilaçlarla tedavi edilebilir. Nöbetler mevcut olduğunda, standart antiepileptik ilaçlarla kontrol edilebilir. Hipotiroidizm tedavisi yararlı görünmemektedir.
Hastalıkla İlgili Genler
AHDS, tiroid hormonu T3 için spesifik bir taşıyıcı olan monokarboksilat taşıyıcı 8’i (MCT8) kodlayan SLC16A2 genindeki (Xq13.2) mutasyonlardan kaynaklanır . Nörolojik sorunlar, tiroid hormonu T3’ün bazı nöronal hücrelere taşınamamasından kaynaklanabilir.
Prognoz
Birkaç hasta 60’larında hayatta kalmasına rağmen, çoğu hasta bağımsız olarak oturamadığı, ayakta duramadığı veya yürüyemediği için genel yaşam beklentisi tehlikeye giriyor ve yaşam kalitesi ciddi şekilde etkileniyor.
Diğer İsimleri
- AHDS
- Allan-Herndon sendromu
- Monokarboksilat taşıyıcı-8 eksikliği
- Triiyodotironin direnci
- T3 direnci
- Zihinsel yetersizlik ve kas atrofisi
- Hipotoni ile X’e bağlı zihinsel engel
Kaynakça
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=1315&Disease_Disease_Search_diseaseGroup=ALLAN—HERNDON—DUDLEY-Syndrome&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Allan-Herndon-Dudley-syndrome&title=Allan-Herndon-Dudley%20syndrome&search=Disease_Search_Simple
- https://ghr.nlm.nih.gov/condition/allan-herndon-dudley-syndrome#
- https://omim.org/entry/300523?search=allan%20herndon%20dudley%20syndrome&highlight=%28syndrome%7Csyndromic%29%20allan%20dudley%20herndon
- https://rarediseases.org/gard-rare-disease/5617/allan-herndon-dudley-syndrome/
- https://www.researchgate.net/figure/Facial-appearance-of-a-female-with-Alan-Herndon-Dudley-syndrome-AHDS-and-de-novo_fig1_5455175