Genel Bilgi
Alfa-mannosidoz (α-mannosidoz), lizozomal α-D-mannosidaz enzimini kodlayan gendeki mutasyonların neden olduğu otozomal resesif kalıtım ile seyrek görülen bir lizozomal depo hastalığıdır. Zihinsel yetersizlik, işitme kaybı, ataksi, iskelet anormallikleri ve anormal yüz özellikleri ile karakterizedir. Belirti ve semptomlar hastalığın şiddetine göre değişiklik gösterebilir. Genellikle hafif ila orta derecede zihinsel engel, işitme kaybı, zayıflamış bağışıklık sistemi, ayırt edici yüz özellikleri ve serebellar bozukluklar (örn. , ataksi) görülür.
Belirti ve Semptomlar
Alfa-mannosidozun semptomları, ilerlemesi ve şiddeti, aynı mutasyonu paylaşan kardeşler dahil olmak üzere bir kişiden diğerine büyük ölçüde değişiklik gösterir. Bazı bireylerde doğumdan kısa bir süre sonra semptomlar gelişebilir. Bebeklik veya erken çocukluk döneminde potansiyel olarak hayatı tehdit eden komplikasyonlar gelişebilir. Diğer bireylerde ise genellikle 10 yaşından önce başlayan daha ılımlı semptomlar gelişebilir. Bazı durumlarda ise bireyler yetişkinliğe kadar teşhis edilemeyebilir. Genel olarak, etkilenen bireyler zihinsel engel, ayırt edici yüz özellikleri ve iskelet anormalliklerine sahip olabilir. Semptomlar zamanla yavaşça kötüleşebilir.
Semptomların şiddetine göre 3 alt tipte sınıflandırılır:
Tip 1: Kas problemleri (miyopati) ile yavaş ilerlemenin olduğu on yaşından sonra gelişen semptomları içeren hafif bir formdur.
Tip 2: İskelet anormallikleri, miyopati ve yavaş ilerleme ile birlikte on yaşından önce semptomların görüldüğü ılımlı bir formdur. İskelet anormallikleri genellikle azalmış kemik yoğunluğu, kafatasının üstündeki kemiklerin kalınlaşması, omurgadaki kemiklerin deformasyonları, eğilmiş bacaklar, kemik ve eklemlerde deformasyonları içerir. En yaygın olan tiptir.
Tip 3: Progresif santral sinir sistemi tutulumu ve enfeksiyonundan dolayı gebelik kaybı veya erken ölüm olarak kendini gösteren şiddetli bir formdur.
Karakteristik yüz özellikleri arasında büyük bir kafa, belirgin alın, düşük saç çizgisi, yuvarlak kaşlar, büyük kulaklar, burnun düzleştirilmiş köprüsü, çıkıntılı çene, geniş aralıklı dişler, büyümüş diş etleri ve büyük dil sayılabilir.

Şekil 1. Alfa-mannosidozda yüz özellikleri. A. 18 aylık ikizler. Genişlemiş baş, kısa boyun, yuvarlak kaşlar, yayılmış burun ve belirgin alın görülmektedir. B. Aynı ikizler 8 yaşında. Ellerin hafif kas atrofisi dikkat çeker. C. İşitme cihazı kullanan 27 yaşındaki alfa-mannosidoz hastası.
Diğer belirtiler arasında şunlar olabilir:
- Hareketleri koordine etmede zorluk (ataksi)
- Oturma ve yürüme gibi motor becerilerinde gecikme
- Konuşma bozuklukları
- İmmün yetmezlik nedeniyle artan enfeksiyon riski
- Karaciğer ve dalağın genişlemesi (hepatosplenomegali)
- Beyindeki sıvı birikmesi (hidrosefali)
- İşitme kaybı
- Göz merceğinin bulanıklaşması (katarakt) ve yakını görememe gibi göz problemleri
- Eklem ağrısı ve iltihabı
- Alfa-mannosidozu olan bazı kişilerin depresyon, anksiyete veya halüsinasyonlar gibi zihinsel sorunları vardır. Kalp ve böbrek problemleri de ortaya çıkabilir.
Prevalans
Alfa-mannosidozun prevalansı hakkında çok az şey bilinmektedir. Avustralya’da yapılan bir araştırmada, 500,000 kişide bir yaygınlık olduğu bildirilmiştir. Norveç’te yapılan bir çalışmada 4.5 milyonluk bir nüfusta sadece altı kişide bu hastalığın varlığı bildirilmiştir. Ayrıca Çekya’da 300,000 kişide bir kişi olarak bildirilmiştir. Hastalık herhangi bir etnik gruba özgü değildir; dünyanın her yerinden bireylerde tanımlanmıştır.
Alfa-mannosidozun ayrıca dünya çapında yaklaşık 500,000 kişiden birinde meydana geldiği tahmin edilmektedir.
Kalıtım Paterni
MAN2B1 genindeki mutasyonlar alfa-mannosidoza neden olur. Bu gen, alfa-mannosidaz enzimini yapmak için talimatlar sağlar. Bu enzim lizozomlarda çalışır. Enzim lizozomlar içinde, belirli proteinlere (glikoproteinler) bağlı oligosakkaritlerin parçalanmasına yardımcı olur. Özellikle, alfa-mannosidaz enzimi mannoz adı verilen şeker molekülü içeren oligosakkaritlerin parçalanmasına yardımcı olur.
MAN2B1 genindeki mutasyonlar, alfa-mannosidaz enziminin mannoz içeren oligosakkaritlerin parçalanmasındaki rolünü yerine getirme kabiliyetine müdahale eder. Bu oligosakkaritler lizozomlarda birikir ve hücrelerin arızalanmasına ve sonunda ölmesine neden olur.
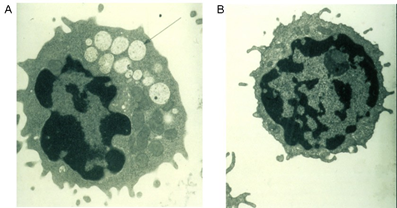
Şekil 2. Oligosakkarit birikimi sonucu koful oluşturan bir lenfosit ve normal lenfosit elektron mikrografisi. A. Alfa-mannosidoz hastasında koful oluşturan bir lenfosit. B. Normal olan bir lenfosit.
Dokular ve organlar, oligosakkaritlerin anormal birikimi ve ortaya çıkan hücre ölümü nedeniyle hasar görür ve alfa-mannosidozun karakteristik özelliklerine yol açar.
Teşhis Yöntemleri Ve Tedaviler
Lizozomal depo hastalığı olan alfa-mannosidozdaki ana klinik özellikler diğer lizozomal depo hastalıkları ile örtüşebilir. Bununla birlikte, bu diğer lizozomal depo hastalıkları ile ilişkili ayırt edici klinik özellikler, klinik laboratuvarlarda biyokimyasal testlerin mevcudiyeti ve doğal geçmişlerinin anlaşılması, bunların birbirinden ayırt edilmesine yardımcı olmaktadır.
Alfa-mannosidoz ön tanısı konulan bir kişide hastalığın ve hastalık için gerekli kişisel tedavilerin belirlenmesi için, aşağıdaki değerlendirmeler önerilir:
- Hastanın işitme kaybı, sinirlilik, depresyon; sosyal, ev, okul veya işle ilgili aktivitelerde veya yürüme mesafesindeki değişiklik; ishal veya idrar tutamama, kas ağrısı, eklem ağrısı, azalmış hareket aralığı ve kemik ağrısı gibi şikayetlerinin tıbbi öyküsünün belirlenmesi gerekmektedir.
- Otoskopi, oftalmoskopi, karaciğer ve dalak boyutunun değerlendirilmesi, kalp ve akciğerlerin oskültasyonu, yürüyüş dahil nörolojik durum ve eklem hareket açıklığının ortopedik değerlendirmesini içeren fizik muayene yapılmalıdır. Çocuklarda standart büyüme çizelgeleri kullanarak boy, ağırlık ve özellikle baş çevresinin büyüklüğü ölçülüp büyüme değerlendirilmelidir.
- İşitme bozukluğu ve orta kulak enfeksiyonlarını tespit etmek için bir kulak burun boğaz uzmanı tarafından muayene edilmelidir.
- Kornea opasiteleri, miyopi, hipermetropi ve şaşılık açısından oftalmolojik olarakdeğerlendirilmelidir.
- Fonksiyonel seviyeyi ve öğrenme kapasitesini belirlemek için nöropsikolojik testler uygulanmalıdır.
- Sistemik lupus eritematozusu (SLE) hariç tutmak için klinik muayene ve immünolojik testler (örn., Antinükleer antikorlar, anti-ds-DNA antikorları) ile kan testleri yapılmalıdır.
- Başın düz radyografileri, dizler (ön-arka görünüm), omurga (yan görünüm) ve herhangi bir semptomatik bölgenin iskelet değerlendirmesi yapılmalıdır.
- Yaşlı bireylerde osteopeni veya osteoporozu tespit etmek için kemik dansitometrisi uygulanmalıdır.
- Özellikle hidrosefali belirtileri ve semptomları varsa (örneğin, baş ağrısı, artan yürüyüş ataksisi, bulantı) ventriküllerin boyutunun ve serebellumun şeklinin ve boyutunun değerlendirilmesi için beynin BT taraması yapılmalıdır.
- Bir klinik genetik uzmanı ve / veya genetik danışman ile konsültasyon.
Çok semptomatik bir hastalığın karakteristik bulgularının tanımlanması üzerine aşağıda açıklanan testlerin sonuçlarına dayanarak alfa-mannosidoz tanısı konulmasına yardımcı olunabilir.
İdrarda oligosakkaritler: İdrardaki mannoz açısından zengin oligosakkarit konsantrasyonlarını ölçmek için bir ön araştırma yapılabilir. Mannoz zengini oligosakkaritlerin yüksek idrar atılımı düşündürücüdür, ancak hastalığın tanısı için yeterli değildir.
Alfa-mannosidaz aktivitesi: Tanı, bir florometrik analiz yoluyla lökositlerde veya diğer çekirdekli hücrelerdeki alfa-mannosidaz aktivitesinin ölçülmesi ile doğrulanır. Bu test, genetik testle birlikte en güvenilir tanı yöntemidir.
Genetik test: Hastalığa neden olan mutasyonun, periferik kan örneği kullanılarak polimeraz zincir reaksiyonu (PCR) amplifikasyonu ile tanımlanması güvenilir bir tanı yöntemidir.
Uygulanan Tedaviler
Konjenital alfa-mannosidoz için bir tedavi yoktur ve genel olarak, ortaya çıkan komplikasyonları önlemek amacıyla hastalığın yönetimi ele alınır. Aşılar, antibiyotikler, işitme cihazları, gözlükler, ortopedik ve diğer yardımcı cihazlar, eğitim müdahaleleri ve konuşma terapisi gibi bireysel semptomlara yönelik tedaviler gerektiği şekilde önerilir. Sağlığı ve tedaviye yanıtı izlemek için düzenli takip önerilmektedir.
Araştırılmakta Olan Tedaviler
Kemik iliği nakli
Alfa-mannosidoz tedavisinde kemik iliği nakli denenmiştir. Kemik iliği nakli ile erken tanı ve hızlı tedavi, bilişsel gerilemeyi önleme ve semptomları iyileştirme şansını arttırır. Bununla birlikte, bu potansiyel tedavinin uzun vadeli güvenliğini ve etkinliğini belirlemek için daha fazla araştırmaya ihtiyaç vardır. Prosedürü ciddi komplikasyon riski taşır. Kemik iliği naklinden sonra, normal gelişim sağlanamamasına rağmen, etkilenen bireyler gelişimsel ilerleme kaydetmiştir. En iyi donör ailesel HLA-özdeş olandır, ancak çoğu zaman bu tip donör tanımlanamaz, bu durumda enzim replasman tedavisi (ERT) en iyi seçenek olarak belirlenebilir.
Gen Terapisi
Gen terapisi ayrıca bazı lizozomal depo bozuklukları için olası bir tedavi olarak araştırılmaktadır. Aktif enzim üretebilen normal genin kalıcı transferi göz önüne alındığında, bu tedavi şeklinin teorik olarak bir tedaviye yol açması muhtemeldir. Bununla birlikte, şu anda, gen terapisi başarılı olmadan önce çözülmesi gereken birçok teknik zorluk vardır.
Hastalıkla İlişkili Genler
MAN2B1geni
Hastalığın Diğer İsimleri
Alfa-D-mannosidoz
Alfa-mannosidaz B eksikliği
Alfa-mannosidaz eksikliği
Lizozomal alfa B mannosidoz
Lizozomal alfa-D-mannosidaz eksikliği
Mannosidoz
Kaynaklar
- https://www.ncbi.nlm.nih.gov/books/NBK1396/
- https://ghr.nlm.nih.gov/condition/alpha-mannosidosis#
- https://www.omim.org/entry/248500?search=Alpha-mannosidosis&highlight=%22alpha%20mannosidosi%22%20%22alpha%7Cmannosidosi%22%20%28alphamannosidosi%7C%20%29%20alphamannosidosi#populationGenetics
- https://rarediseases.org/rare-diseases/alpha-mannosidosis/
- https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-3-21