Genel Bilgi
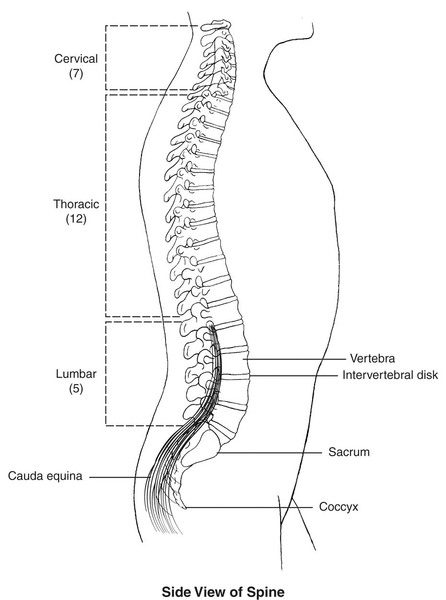
Schimke immüno-ossöz displazisi , kısa boy, böbrek hastalığı ve zayıf bir bağışıklık sistemi ile karakterize bir durumdur. Bu durumu olan insanlarda, kısa boy düzleşmiş omurga kemiklerinden ( vertebralar) kaynaklanır.), Görsel 1 kısaltılmış boyun ve gövde ile sonuçlanır. Yetişkin yüksekliği tipik olarak 3 ila 5 feet arasındadır. Böbrek (böbrek) hastalığı sıklıkla hayatı tehdit eden böbrek yetmezliğine ve son dönem böbrek hastalığına (ESRD) yol açar. Etkilenen bireyler ayrıca T hücreleri adı verilen bazı bağışıklık sistemi hücrelerinin yetersizliğine de sahiptir.. T hücreleri yabancı maddeleri tanımlar ve vücudu enfeksiyonlara karşı korur. T hücrelerinin yetersizliği, bir kişinin hastalığa karşı daha duyarlı olmasına neden olur.
Bu rahatsızlığı olan kişilerde sık görülen diğer özellikler arasında alt sırtın abartılı bir eğriliği vardır ( lordoz); tipik olarak göğüste ve sırtta koyulaşmış cilt lekeleri (hiperpigmentasyon); ve burnun yuvarlatılmış ucu olan geniş bir burun köprüsü.
Schimke immüno-ossöz displazinin daha az görülen belirtileri ve semptomları , atardamarların astarında ( ateroskleroz) yağ birikintileri ve skar benzeri bir doku birikimini içerir.) beyine kan akışını azalttı (beyin iskemisi), migren benzeri baş ağrıları, yetersiz tiroit bezi (hipotiroidizm), azalmış beyaz kan hücreleri (lenfopeni), az gelişmiş kalça kemikleri (hipoplastik pelvis), anormal derecede küçük baş büyüklüğü ( mikrosefali)), erkeklerde sperm eksikliği (azospermi) ve kadınlarda düzensiz adet kanaması.
Şiddetli vakalarda, doğumda Schimke immüno-ossöz displazinin birçok belirtisi mevcut olabilir. Bu hastalığın hafif vakaları olan kişiler geç çocukluğa kadar belirti veya semptom geliştirmeyebilir.
Schimke immuno-ossöz displazili hastaların yaklaşık yarısında SMARCAL1 geninde mutasyon tanımlanmamıştır. Bu gibi durumlarda, hastalığın nedeni bilinmemektedir.
Görsel 1, Kaynak: https://ghr.nlm.nih.gov/art/large/normal-spine.jpeg
{kind=link}
Bazı insanlar erken çocukluk döneminde şiddetli bir form geliştirirken, diğerleri çocuklukta veya sonrasında hafif bir form geliştirir. Kısa boy, omurganın anormal gelişimini ve uzun kemiklerin uçlarını içeren spondiloepifizeal displaziden kaynaklanır . Neredeyse SIOD’lu tüm kişilerin böbrek hastalığı vardır, bu da böbrek hastalığına son aşamada ilerler.
Schimke immünoosöz displazisi (SIOD) şiddetine göre değişmektedir. Erken başlangıç formuna sahip olanlar genellikle ciddi semptomlara ve yaklaşık 9 yıllık ortalama ömre sahiptir. Ölüm nedenleri inme, ciddi fırsatçı enfeksiyon, kemik iliği yetmezliği, böbrek yetmezliği komplikasyonları, konjestif kalp yetmezliği veya akciğer hastalığını içerebilir. Daha hafif belirtileri olanlarda yetişkinlik çağında hayatta kalırlar, böbrek hastalığı iyi yönetilerek. Bununla birlikte, başlangıç şiddeti ve yaşının mutlaka yaşam beklentisini kesin göstermeyeceğini not etmek önemlidir, çünkü şiddetli, erken başlangıçlı SIOD’lu bazı insanlar 20’li ve 30’lu yaşlarında hayatta kalmıştır.
Genetik Değişiklikler /Etken Faktörler
SMARCAL1 genindeki mutasyonlar, Schimke immüno-osöz displazisi riskini arttırır. SMARCAL1 gen olan spesifik fonksiyonu bilinmeyen bir protein üretmek için talimatlar sağlar. SMARCAL1 proteini kromatine bağlanabilir DNA’yı kromozomlara paketleyen DNA ve protein kompleksidir. Benzer proteinlerin fonksiyonuna dayanarak, SMARCAL1’in, kromatin remodeling olarak bilinen bir işlemle diğer genlerin aktivitesini (ekspresyonunu) etkilediği düşünülmektedir. Kromatinin yapısı, DNA’nın ne kadar sıkı bir şekilde paketlendiğini değiştirmek için değiştirilebilir (yeniden yapılandırılabilir). Kromatin remodeling, gelişim sırasında regresyonun düzenlenmesinin bir yoludur. DNA sıkıca paketlendiğinde, gen ekspresyonu, DNA’nın gevşek bir şekilde paketlendiğinden daha düşüktür.
SMARCAL1 genindeki mutasyonların, protein aktivitesini, protein stabilitesini veya proteinin kromatine bağlanma yeteneğini etkileyerek hastalığa yol açtığı düşünülmektedir. SMARCAL1 genindeki mutasyonların kromatin remodelingine ve diğer genlerin ekspresyonuna müdahale edip etmediği açık değildir.
Schimke immüno-disöz displazisi ile ilişkili mutasyonlar, SMARCAL1 proteininin olağan fonksiyonlarını bozar veya herhangi bir fonksiyonel proteinin üretimini önler. Fonksiyonel bir protein eksikliğine neden olan mutasyonlara sahip olan insanlar, aktif fakat hatalı çalışan bir proteine yol açan mutasyonlara sahip olanlardan daha şiddetli bir şekilde bu hastalığa sahip olma eğilimindedir. Bununla birlikte, SMARCAL1 gen mutasyonlarına sahip kişilerin Schimke immüno-kemikli displazisi geliştirmesi için , şu anda bilinmeyen diğer genetik veya çevresel faktörlerin de bulunması gerekir.
Belirti ve Semptomlar
Schimke
immünoosöz displazisi (SIOD) vücudun birçok bölümünü etkiler. Yavaş büyüme
genellikle SIOD’un ilk işaretidir. İnsanlar genellikle kısa bir boyun ve
gövde geliştirir (orantısız şekilde).kısa
boy) spondiloepifizeal displazi nedeniyle . Kemik
anormallikleri tipik olarak omurga veya kalçalarda gelişir. SIOD’lu birçok
kişi sonunda kalça
protezi ameliyatına ihtiyaç
duyar .Böbrek hastalığı tanı anında var olur veya birkaç
yıl içinde gelişir ve sonuçta böbrek yetmezliğine ilerler . Neredeyse
tüm SIOD’lu kişilerin kanı varhücreEn sık T-hücrelerinin
eksikliği. Bu, hayati tehlike oluşturabilecek enfeksiyon riskinin
artmasına neden olur.
Diğer belirti ve semptomlar şunları
içerebilir:
- Azaltılmış tiroid fonksiyonu
- Çıkıntılı karın
- Cilt renginin artması yamalar (hiperpigmente maküller)
- Erken çocukluk döneminde başlayan ilerleyici ateroskleroz
- Geçici iskemik ataklar veya inme
- Şiddetli, migren benzeri baş ağrıları
- Pulmoner emboli , pulmoner hipertansiyon ve akciğer hastalığı dahil olmak üzere akciğer komplikasyonları
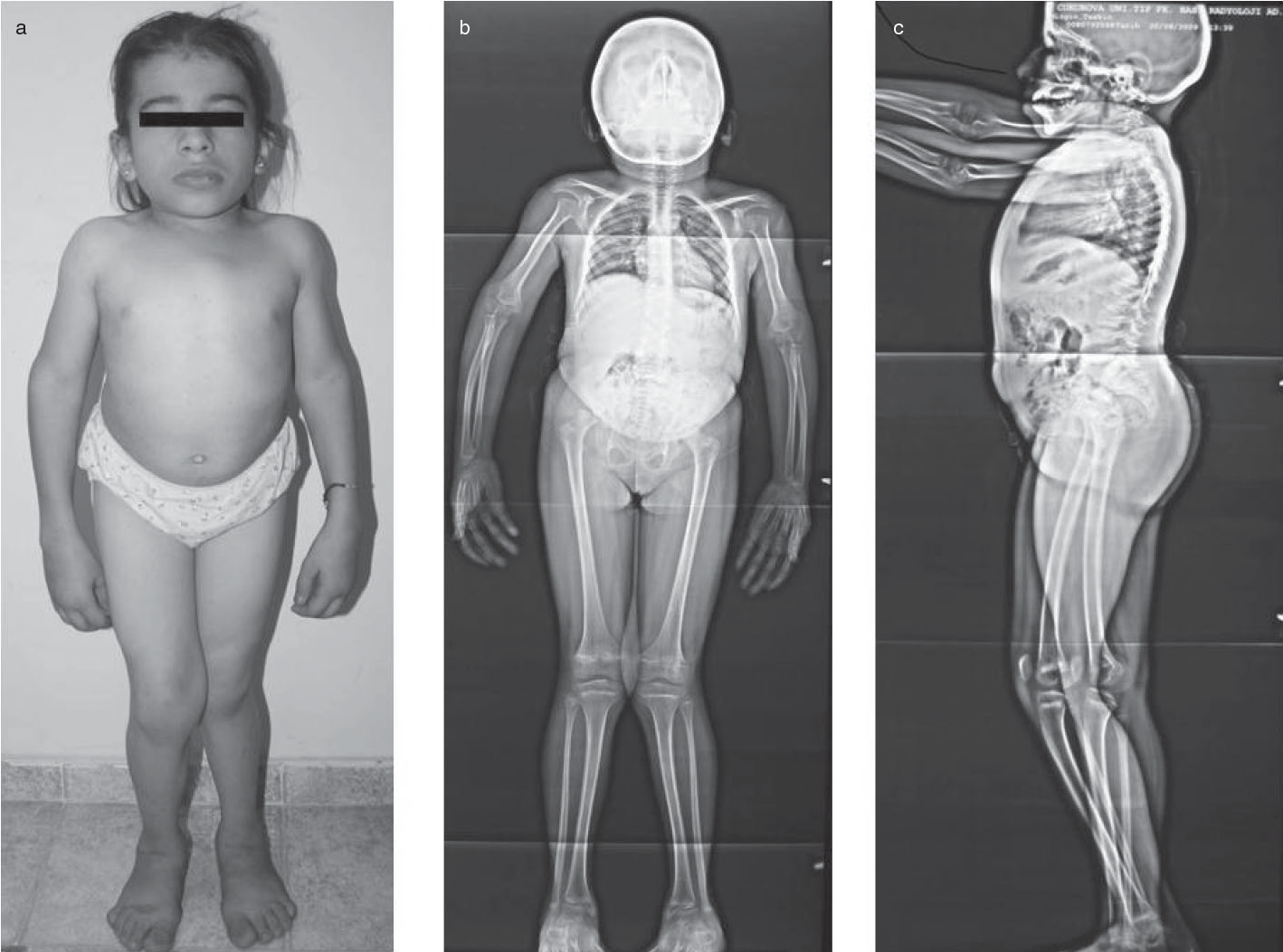
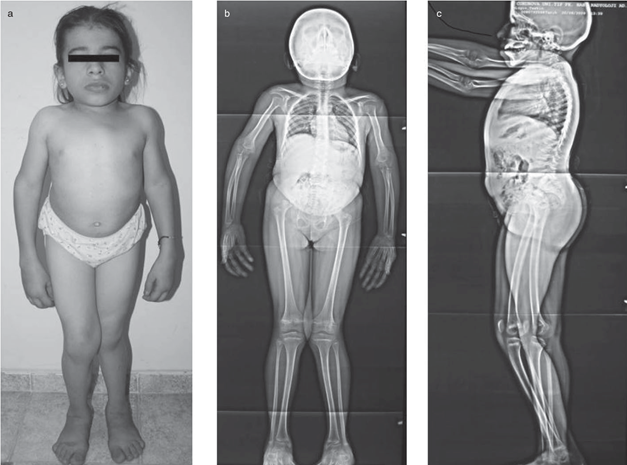
Görsel 2: kaynak: (a) Üçgen yüz, geniş burun köprüsü, soğanlı burun ucu, kısa boyun ve gövde ile kısa boy, bel lordozu ve çıkıntılı karın gösteren kısa fotoğraflar. (b) Postero-ön ve (c) yanal X-ışınları gösteriyor https://d3i71xaburhd42.cloudfront.net/48a0aa9394a5ded8787b9bf98fb8c19677ca0600/2-Figure1-1.png
{kind=link}
Genetik Görülme Sıklığı
Schimke immuno-osseöz displazisi çok nadir görülen bir durumdur. Kuzey Amerika’da yaygınlığın 1 milyondan 3 milyona kadar bir insan olduğu tahmin edilmektedir.
Kalıtım Paterni\Deseni
SMARCAL1 genindeki mutasyonlar, otozomal resesif paternde kalıtsaldır bu, Schimke immüno-ossöz displazisi riskinin artmasının, her hücrede SMARCAL1 geninin her iki kopyasındaki mutasyonlardan kaynaklandığı anlamına gelir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve semptomlarını göstermezler.
Kavramsal olarak, etkilenen bir bireyin her birinin % 25’i etkilenme şansı, %50’sinin asemptomatik bir taşıyıcı olma olasılığı ve % 25’inin de etkilenmeme ve taşıyıcı olmama olasılığı vardır. Ailede patojenik varyantların her ikisi de biliniyorsa taşıyıcı testi ve doğum öncesi testi mümkündür.
Teşhis Yöntemleri ve Tedavileri
Teşhis:
Aşağıdaki temel özelliklere sahip kişilerde SIOD tanısından şüphelenilir:
- Kısa boy
- Spondiloepifizal displazi
- ilerici böbrek hastalığı
- T-hücre eksiklik
- Karakteristik yüz özellikleri
- Cilt renginin artması yamalar (hiperpigmente maküller)
Tanı klinik muayeneden sonra konulabilir. Genetik test tespit etmek mutasyonlariçinde SMARCAL1 genteşhisi onaylayabilir.
Kıkırdak kılı hipoplazisi (bu terime bakınız) ana ayırıcı tanıdır.
Tedavi:
Schimke
immunoosseöz displazinin (SIOD) tedavisi her bireyin şiddetine ve bireysel
semptomlarına bağlıdır. Kalça, böbreklerin düzenli olarak izlenmesi,bağışıklık sistemive kan tavsiye edilir.
SIOD için gerekli olabilecek tedavi örnekleri
şunları içerir:
- Böbrek diyaliz veya nakli
- Kalça protezi
- Nötropeninin granülosit ile tedavisi koloni uyarıcı faktör veya granülosit-makrofaj koloni uyarıcı faktör
- Kemik iliği nakli immün yetmezlik için (transplantasyon sonrası birkaç ölüm bildirilmiş olmasına rağmen)
- Otoimmün semptomları olanlar için bağışıklık sistemini baskılayan ilaçlar
- Tekrarlayan herpes enfeksiyonları için asiklovir
- Şiddetli papilloma tedavisinde Imiquimod ve cidofovir (bir antiviral)virüs cilt enfeksiyonları
- Geçici iskemik atakları (“mini vuruş”) veya felç tedavisinde kan akışını iyileştiren veya kanın pıhtılaşmasını azaltan ajanlar
- Hipotiroidizmin standart tedavisi
- Standart tedavi skolyoz
Hastalıkla İlişkili Genler
Schimke immüno-ossöz displazisi (SIOD), spondiloepifizeal displazi ve orantısız kısa boy, yüz dismorfizmi, T hücreli immün yetmezlik ve nefrotik sendromlu glomerülonefrit ile karakterize multisistemik bir hastalıktır.
SIOD , SMATCAL1 genindeki (2q35), kromatin remodeling proteini hHARP’yi (ayrıca kromatin alt-ailesi A-benzeri protein 1’in SWI / SNF ile ilgili matris-ilişkili aktin bağımlı regülatörü olarak da bilinir) kodlayan mutasyonlardan kaynaklanır .
Hastalığın Diğer İsimleri
- immüno-displazik, Schimke tipi
- Schimke immünoosöz displazisi
- SIOD
- Spondiloepifizal displazi nefrotik sendrom
- Schimke sendromu;
- İmmünoosöz displazi, Schimke tipi
Kaynaklar
- https://ghr.nlm.nih.gov/condition/schimke-immuno-osseous-dysplasia
- https://rarediseases.info.nih.gov/diseases/4984/schimke-immunoosseous-dysplasia
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=1830
- https://www.ncbi.nlm.nih.gov/books/NBK1376/
- https://www.semanticscholar.org/paper/Medullary-nephrocalcinosis-in-Schimke-dysplasia.-Yavuz-Bayazit/48a0aa9394a5ded8787b9bf98fb8c19677ca0600 (Görsel 2’nin kaynağı)