Turner sendromu, kızlar arasında en çok görülen kromozomal anomalilerden biridir. Nedeni, dişilerde 2 adet olması gereken X kromozomlarından birinin yokluğu ya da anormal olmasıdır. En tipik belirtileri, geniş ya da yele (perdeli) boyun (ekstra deri kıvrımları), bebeklerde şişmiş el ve ayaklar (lenfödem) , düz ve dışa dönük tırnaklar, iskelet anormallikleri veya böbrek problemleri vardır. Turner sendromlu bireylerin üçte biri ila yarısı, kalpten çıkan büyük arterin daralması (aort koarktasyonu) veya aortu kalbe (aort kapak) bağlayan kapak anormallikleri gibi bir kalp kusuru ile doğar. Bu kalp kusurları ile ilişkili komplikasyonlar hayatı tehdit edici olabilir. Turner sendromunun en yaygın özelliği, yaklaşık 5 yaşlarında ortaya çıkan kısa boydur. Erken yumurtalık fonksiyon kaybı (yumurtalık hipofonksiyonu veya erken yumurtalık yetmezliği) de çok yaygındır. Yumurtalıklar ilk başta normal olarak gelişir, ancak yumurta hücreleri (oositler) genellikle erken ölür ve çoğu yumurtalık dokusu doğumdan önce dejenere olur. Turner sendromlu çoğu kız ve kadın normal zekaya sahiptir. Bu özellikler etkilenen bireyler arasında farklılık gösterse de, gelişimsel gecikmeler, sözsüz öğrenme güçlükleri ve davranış problemleri mümkündür.
TS ilk olarak 1938’de Amerika Birleşik Devletleri’nde Dr. Henry Turner tarafından tanımlanmıştır.
Genetik Değişiklikler /Etken Faktörler
Turner sendromu, iki cinsiyet kromozomundan biri olan X kromozomu ile ilişkilidir. İnsanlar tipik olarak her hücrede iki cinsiyet kromozomuna sahiptir: dişilerde iki X kromozomu bulunurken, erkeklerde bir X kromozomu ve bir Y kromozomu bulunur. Turner Sendromu, bir dişi hücrelerinde normal bir X kromozomu bulunduğunda ve diğer cinsiyet kromozomu eksik olduğunda veya yapısal olarak değiştiğinde ortaya çıkar. Eksik genetik materyal doğumdan önce ve sonra gelişimi etkiler.
Turner sendromlu bireylerin yaklaşık yarısında monozomi X vardır, bu da bireyin vücudundaki her hücrenin normal iki cinsiyet kromozomu yerine X kromozomunun yalnızca bir kopyasına sahip olduğu anlamına gelir.
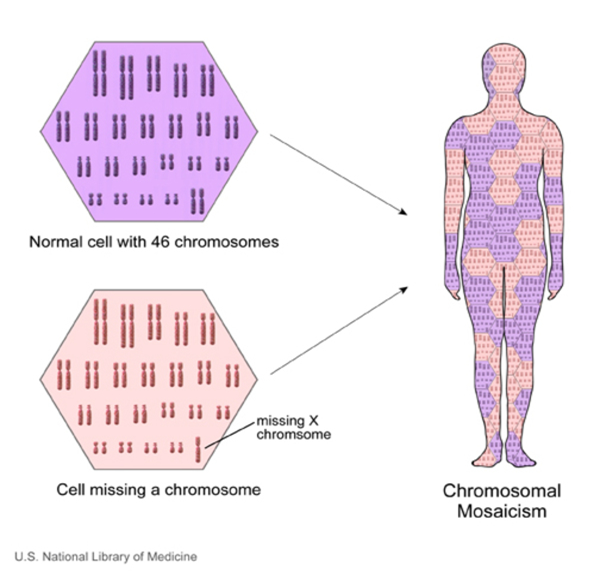
Turner sendromlu bazı kadınlar, hücrelerinin sadece bazılarında mozaikçilik olarak bilinen kromozomal bir değişikliğe sahiptir. X kromozomu mozaikçiliğinin neden olduğu Turner sendromlu kadınların mozaik Turner sendromuna sahip olduğu söylenir.
Belirti ve Semptomlar
Görsel 1: https://media.sciencephoto.com/image/c0328634/800wm

Görsel 2, kaynak: https://www.medicalhomeportal.org/image/401

Turner sendromunun semptomları ve şiddeti bir kişiden diğerine oldukça değişebilir. Bozukluğun birçok özelliği spesifik değildir ve diğerleri zamanla yavaşça gelişebilir veya daha az belirgin olabilir. Etkilenen bireylerin aşağıda tartışılan semptomların hepsine sahip olmayabileceğini belirtmek önemlidir. Etkilenen bireyler, özel vakaları, ilişkili semptomları ve genel prognozu hakkında doktorları ve tıbbi ekipleriyle konuşmalıdır.
Turner sendromlu hemen hemen tüm kadınlar büyüme başarısızlığı gösterir ve ortalamadan daha kısa bir son boyuta (kısa boy) ulaşır. Çocuklar başlangıçta, genellikle yaşamın ilk birkaç yılında normal büyüme gösterebilir. Bununla birlikte, çoğu durumda, büyüme oranı nihayetinde normalden daha yavaş olur ve etkilenen çocuklar normal büyüme atakları yaşamazlar (örneğin ergenlik döneminde büyüme fışkırması olmaz).
Turner sendromunun bir diğer yaygın özelliği, yumurtalıkların düzgün gelişememesidir (gonadal disgenez). Gonadal disgenez, çocukluk döneminde erken yumurtalık fonksiyonunun kaybına neden olabilir (erken yumurtalık yetmezliği). Normal olarak, yumurtalıklar ergenlikte seks hormonları (örn. Östrojen ve progesteron) üretir. Bu hormonlar ergenliğin başlaması ve ikincil cinsel özelliklerin doğru gelişimi için gereklidir. Etkilenen kadınların çoğu, göğüsleri ve normal kadın vücudu hatlarını geliştirmek, uygun kemik büyümesine girmek ve adet görmeye başlamak için hormon replasman tedavisine ihtiyaç duyacaktır. Bazı durumlarda, etkilenen bireyler meme gelişimine girebilir ve terapi olmadan menstruasyona başlayabilir (spontan pubertal gelişim), ancak çoğu cinsel gelişimini durduracak ve gençlik yıllarında daha sonraki bir noktada adet görmeyi durduracaktır.
Turner sendromlu dişiler, perdeli bir görünüme sahip kısa bir boyun, başın arkasındaki düşük bir saç çizgisi, düşük ayarlı kulaklar ve yukarı doğru çevrilmiş dar tırnaklar ve ayak tırnakları gibi çeşitli farklı fiziksel özellikler geliştirebilir. Bazen “kalkan sandığı” olarak adlandırılan geniş aralıklı meme uçlarına sahip geniş bir göğüs oluşabilir. Bazı bireylerin şişmiş, kabarık elleri ve ayakları olabilir. Bu belirtiler, lenfatik sistemi etkileyen bir durum olan lenfödem nedeniyle ortaya çıkabilir. Lenfatik sistem, vücutta belirli protein açısından zengin sıvı (lenf) ve kan hücrelerini filtreleyen ve dağıtan damarlar, kanallar ve düğümlerin dolaşım ağıdır. Lenfödem, vücudun etkilenen bölgelerindeki sıvı birikimine (ödem) bağlı şişme ile karakterizedir.
Ek fiziksel bulgular arasında bir çene (retrognati), çapraz gözler (şaşılık), tembel gözler (ambliyopi), sarkık göz kapakları ve ağızda darlık, yüksek kemerli bir çatı (damak) sayılabilir. Bazı bireylerde ellerin kısa kemikleri, özellikle dördüncü metakarplar, dirseklerde ortaya çıkan kollar ve düz ayaklar (pes planus) dahil iskelet malformasyonları olabilir. Olguların yaklaşık% 10’unda, omurganın anormal yanal eğriliği (skolyoz) de ortaya çıkabilir.
Bazı Turner sendromu vakalarıyla ilişkili kalp kusurları, akciğerlerin arterlerinin yüksek tansiyonu (pulmoner hipertansiyon) veya içte bir yırtık olan aort diseksiyonu da dahil olmak üzere ciddi, hayatı tehdit eden komplikasyon riskini artırabilir.
Genetik Görülme Sıklığı
Bu durum dünya çapında 2.500 yeni doğan kızdan yaklaşık 1’inde görülür, ancak terimde hayatta kalmayan hamileliklerde (düşükler ve ölü doğumlar) çok daha yaygındır. Bozukluğun sıklığını etkileyen bilinen herhangi bir ırksal veya etnik faktör yoktur. Bazı durumlarda, bozukluk doğumdan önce veya doğumdan kısa bir süre sonra teşhis edilir. Bununla birlikte, hafif vakalar yaşamın ilerleyen dönemlerine kadar ve hatta yetişkinlik döneminde bile teşhis edilmeden kalabilir.
Kalıtım Paterni / Deseni
Turner sendromu vakalarının çoğu kalıtsal değildir. Bu durum monozomi X’ten kaynaklandığında, kromozomal anormallik, etkilenen kişinin ebeveyninde üreme hücrelerinin (yumurta ve sperm) oluşumu sırasında rastgele bir olay olarak ortaya çıkar. Ayrılma adı verilen hücre bölünmesindeki bir hata, anormal sayıda kromozomlu üreme hücrelerine neden olabilir. Örneğin, bir yumurta veya sperm hücresi, ayrılmanın bir sonucu olarak bir cinsiyet kromozomunu kaybedebilir. Bu atipik üreme hücrelerinden biri çocuğun genetik yapısına katkıda bulunursa, çocuk her hücrede tek bir X kromozomuna sahip olacak ve diğer cinsiyet kromozomunu kaçırmayacaktır.
Mozaik Turner sendromu da kalıtsal değildir. Etkilenen bir kişide, erken fetal gelişimde hücre bölünmesi sırasında rastgele bir olay olarak ortaya çıkar. Sonuç olarak, etkilenen bir kişinin hücrelerinin bazıları normal iki cinsiyet kromozomuna sahiptir ve diğer hücrelerde X kromozomunun yalnızca bir kopyası bulunur. X kromozom mozaikliği olan kadınlarda diğer cinsiyet kromozom anormallikleri de mümkündür.
Nadiren, X kromozomunun kısmen silinmesinin neden olduğu Turner sendromu bir nesilden diğerine geçebilir.
Mozaik Turner sendrom, Görsel, kaynak: https://ghr.nlm.nih.gov/art/large/mosaicism.jpeg
{kind=link}
Teşhis Yöntemleri ve Tedavileri
Benzer Bozukluklar
Aşağıdaki bozuklukların belirtileri Turner sendromunun semptomlarına benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir.
Noonan sendromu, doğumda tipik olarak görülen (konjenital) yaygın bir genetik bozukluktur. Bozukluk, aralık ve şiddet açısından büyük ölçüde değişen geniş bir semptom yelpazesi ve fiziksel özellik ile karakterizedir. Etkilenen birçok kişide, ilişkili anormallikler ayırt edici bir yüz görünümü içerir; geniş veya perdeli bir boyun; düşük bir posterior saç çizgisi; tipik bir göğüs deformitesi ve kısa boy. Baş ve yüz (kraniyofasiyal) bölgesinin karakteristik anormallikleri, genişçe ayarlanmış gözleri (oküler hipertelorizm); gözlerin iç köşelerini kaplayabilen cilt kıvrımları (efsanevi kıvrımlar); üst göz kapaklarının sarkması (pitoz); küçük bir çene (mikrognati); depresif bir burun kökü; geniş tabanlı kısa bir burun; ve düşük yerleşimli, arkaya döndürülmüş kulaklar (pinnae). Göğüs kemiği (sternum) anormallikleri, omurganın eğriliği (kifoz ve / veya skolyoz) ve dirseklerin dışa doğru sapması (cubitus valgus) gibi belirgin iskelet malformasyonları da tipik olarak mevcuttur. Noonan sendromlu birçok bebekte ayrıca, kalbin sağ alt odasından akciğerlere (pulmoner kapak darlığı) uygun kan akışının engellenmesi gibi kalp (kardiyak) kusurları vardır. Ek anormallikler, belirli kan ve lenf damarlarının malformasyonlarını, kan pıhtılaşmasını ve trombosit eksikliklerini, öğrenme güçlüklerini veya hafif zihinsel özürlülüğü, testislerin etkilenen erkeklerde yaşamın ilk yılına kadar inmesini (ve kriptorşidizmi) içerebilir ve / veya diğer semptom ve bulgular. Noonan sendromu, rasopati yolunu oluşturan çoklu tekli genlerdeki anormalliklerin (mutasyonlar) neden olduğu otozomal dominant bir genetik bozukluktur. Noonan sendromu ile ilişkili bazı semptomlar, Turner sendromu olanlara yüzeysel olarak benzeyebilir (kısa boy, perdeli boyun vb. Gibi her iki bozuklukla ilişkili olabilecek bazı bulgular nedeniyle). Sonuç olarak, geçmişte, Noonan sendromu “erkek Turner sendromu”, “kadın yalancı Turner sendromu” veya “normal kromozomlu karyotipli Turner fenotipi” olarak anılmıştır. Bununla birlikte, iki bozukluk arasında birçok önemli fark vardır. Noonan sendromu hem erkekleri hem de kadınları etkiler ve normal bir kromozomal makyaj (karyotip) vardır. X kromozomunu etkileyen anormallikler ile karakterize edilen Turner sendromundan sadece kadınlar etkilenir.
Teşhis
Ayrıntılı bir hasta öyküsü, kapsamlı bir klinik değerlendirme ve çeşitli özel testler. Büyüme eksikliği veya nedeni bilinmeyen kısa boylu kızlarda Turner sendromundan şüphelenilmelidir.
Turner sendromu tanısı genellikle karyotip belirlenerek elde edilen kromozomal analizle doğrulanır. Karyotipleme, kromozomların sayısını ve yapısını değerlendiren bir laboratuvar testidir.
Bazı durumlarda, fetal ultrasonda Turner sendromu ile ilişkili bazı fiziksel bulgular görülebilir. Örneğin, gelişmekte olan bir fetüsün boynuna yakın lenf sıvısı birikimi bazen rutin bir fetal ultrasonda görülebilir. Kesin test CVS veya amniyosentez ile yapılabilir. CVS, gebeliğin 10-12. haftasında yapılır ve doku örneklerinin plasentanın bir kısmından çıkarılmasını içerirken, amniyosentez 16-18. gebelik haftasında yapılır ve fetüsün etrafında küçük bir sıvı örneği almayı içerir.
Etkilenen bireyleri karaciğer, böbrek veya kalp anormallikleri gibi Turner sendromuyla ilişkili potansiyel semptomların varlığı açısından değerlendirmek için manyetik rezonans görüntüleme (MRI) gibi spesifik görüntüleme teknikleri uygulanabilir.
Tiroid ve karaciğer fonksiyonu, kemik yaşı ve büyüme hakkında ek değerlendirme yapılmalıdır. Hipertansiyon taraması da yapılmalıdır. Doğumda teşhis edilen bebeklere işitme muayenesi de dahil olmak üzere tam bir kulak, burun ve boğaz muayenesi yapılmalıdır. Çocuklar, özellikle tekrarlayan orta kulak iltihabı yaşayanlar ve yetişkinler periyodik işitme değerlendirmesi gerektirir.
Tedavi
Turner sendromunun tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, bir uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, çocuk uzmanları, cerrahlar, kardiyologlar, endokrinologlar, konuşma patologları, kulak burun boğaz uzmanları, göz doktorları, psikologlar ve diğer sağlık uzmanlarının sistematik ve kapsamlı bir şekilde çocuğun tedavisini etkilemesi gerekebilir. Etkilenen bireyler ve aileleri için genetik danışmanlık önerilir.
Belirli ilaç rejimlerinin ve / veya diğer tedavilerin kullanımına ilişkin kararlar, hekim ve sağlık ekibinin diğer üyeleri tarafından, sendromun özelliklerine dayanarak hastayla dikkatli konsültasyonda verilmelidir; olası yan etkiler ve uzun vadeli etkiler de dahil olmak üzere potansiyel faydalar ve riskler hakkında kapsamlı bir tartışma yapılıp; hasta tercihi ve diğer uygun faktörler değerlendirilmelidir.
Turner sendromu için bir tedavi yoktur, ancak fiziksel gelişimi artıracak tedaviler geliştirilmiştir. Uygun tıbbi bakım ile Turner sendromlu kadınlar tam, üretken yaşamlar sürdürebilmelidir. Etkilenen bireyler için birincil tedaviler büyüme hormonu tedavisi ve östrojen tedavisidir. Çocuklardaki büyüme hormonu tedavisi Çocuk endokrinoloji hekimlerince uygun görülen sürede planlanır.
Hormon replasman tedavisi genellikle 12-14 yaşlarında başlar. Çoğu ortalama kız ergenliğe girecek. Ergenliğin başlatılmasının zamanlaması, büyüme hormonu replasmanındaki büyüme ilerlemesini de dikkate alır. Bu özellikleri korumak için replasman tedavisine devam edilmelidir ve çoğu kadın menopoza kadar östrojen ve progesteron tedavisine ihtiyaç duyacaktır.
Ek tedavi semptomatik ve destekleyicidir. Örneğin, tiroid hormonu replasman tedavisi tiroid hastalığı olan bireyleri tedavi etmek için kullanılabilir. İşitme kaybının işitme cihazlarıyla düzeltilmesi, öğrenme ve sosyal etkileşime yardımcı olabilecek bir diğer önemli müdahaledir.
Turner sendromlu çocukların potansiyellerine ulaşmalarını sağlamak için erken müdahale önemlidir.
Tıp uzmanlarının ve iyi bir sosyal destek sisteminin yardımıyla, TS’li bir kadın tatmin edici, sağlıklı bir yaşam sürmeyi bekleyebilir.
Hastalığın Diğer İsimleri
- 45,X
- Monosomy X
- TS
- Turner’s syndrome
- Ullrich-Turner syndrome
Kaynak