Genel Bilgi
L1 sendromu, öncelikle sinir sistemini etkileyen ve neredeyse sadece erkeklerde görülen bir grup durumu tanımlar. Bu koşullar şiddette değişiklik gösterir ve en ağırdan en azına kadar, Sylvius (HSAS) su kemeri stenozu, MASA sendromu, spastik parapleji tip 1 ve X’e bağlı komplike korpus kallusum agenezi olan X’e bağlı hidrosefali içerir.
HSAS, durumun karakteristik özelliklerinin
kısaltmasıdır: beyinde bir sıvı (hidrosefali) genellikle doğumdan önce ortaya
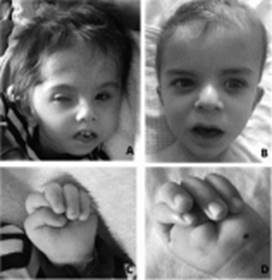
çıkan bir sıvı birikimi, kas sertliği (spastisite), avuç içlerine kalıcı olarak
bükülen baş parmaklar (uyarılmış başparmaklar) ve Beyindeki bir geçidin
daraltılması (darlığı) Sylvius’un su kemeri olarak adlandırılır. HSAS’lı
bireylerde Sylvius’un su kemeri stenozu, ventrikül adı verilen sıvı dolu
boşluklardan beyin omurilik sıvısının (BOS) akışını engelleyerek hidrosefali
oluşturur. HSAS’lı bireyler sıklıkla ciddi zihinsel engellidir ve nöbet
geçirebilirler.
MASA
sendromu ayrıca, hafif ila orta, gecikmeli konuşma (afazi), spastisite ve
eklenmiş başparmaklar arasında değişebilen zihinsel engelli (zihinsel gerilik)
durumun karakteristik özellikleri için de adlandırılmıştır. MASA sendromlu
bireylerde ventrikülde hafif genişleme olabilir.
Spastik
parapleji tip 1, ilerleyici kas sertliği (spastisite) ve uzuvların paralizi gelişimi
(parapleji) ile karakterizedir. Etkilenen bireyler ayrıca hafif ila orta
dereceli zihinsel engellidir. Spastik parapleji tip 1 olan kişilerde genellikle
beyin yapılarında önemli anormallikler yoktur.
X’e
bağlı komplike korpus kallosum agenezisi, beynin sol ve sağ yarısını (korpus
kallosum) bağlayan dokunun az gelişmişliği (hipoplazi) veya yokluğu (agenezisi)
ile tanımlanır. Bu durumu olan insanlar spastik paraplejiye sahip olabilir ve
hafif ila orta derecede zihinsel engelli olabilir.
L1 sendromlu
bireylerin yaşam beklentisi, belirti ve semptomların ciddiyetine bağlı olarak
değişir. Ciddi derecede etkilenen bireyler doğumdan sadece kısa bir süre daha hayatta
kalabilirken, hafif özellikleri olanlar yetişkinlikte yaşar.
L1
sendromunu oluşturan koşulların bir zamanlar farklı bozukluklar olduğu
düşünülüyordu, ancak genetik bir nedeni paylaştığı tespit edildiğinden, şimdi
aynı sendromun bir parçası olarak kabul ediliyorlar. Aynı mutasyonun neden
olduğu L1 sendromlu aile üyeleri, durumun farklı biçimlerine sahip olabilir.
Aşağıda (L1 Sendromlu bir hastada genel problemler) görsel kaynağı: https://www.sciencedirect.com/science/article/pii/S030384671830221X
Genetik Değişiklikler/ Etken Faktörler
L1 sendromuna L1CAM genindeki mutasyonlar neden olur.
L1CAM geni, sinir sistemi boyunca bulunan Ll hücre yapışma molekülü proteinini
(Ll proteinine kısaltılmış) üretmek için talimatlar sağlar. Bu protein,
hücrelerin birbirine yapışmasına yardımcı olmak için komşu nöronlardaki
proteinlere bağlandığı (bağlandığı) sinir hücrelerinin (nöronlar) yüzeyinde
bulunur. L1 proteini beyin gelişimine, düşünme yeteneğine, hafızasına ve
hareketine katkıda bulunan nöronların birçok işlevinde rol oynar.
L1 sendromuna neden olan L1CAM gen mutasyonları, hücre-hücre yapışmasını kolaylaştıramayan veya
çeşitli nöronal fonksiyonlara katılamayan bir L1 proteinine yol açar. Bu
fonksiyonların bozulması muhtemelen beynin büyümesini ve gelişmesini
engelleyerek L1 sendromunun belirti ve semptomlarına yol açar.
Bazı L1CAM gen mutasyonları, anormal derecede kısa ve işlevsel olmayan bir
proteinin üretilmesine veya proteinin tamamen yok olmasına neden olur. Bu
mutasyonlar tipik olarak, sıklıkla HSAS olmak üzere, ciddi L1 sendromu vakalarına
yol açar. Diğer mutasyonlar, proteinin yapısını değiştirir, proteinin hücre
yüzeyindeki diğer proteinlerle etkileşime girme kabiliyetini azaltır veya
proteinin ihtiyaç duyulduğu yerde hücre yüzeyine ulaşmasını önler. Bu
mutasyonlar tipik olarak daha az ciddi L1 sendromu vakalarına, genellikle MASA
sendromuna veya bu durumun diğer hafif formlarına neden olur.
Bir mutasyonun L1 proteini üzerindeki etkisi, bazen durumun ciddiyeti hakkında bir ipucu sunsa da, aynı veya benzer mutasyonlara sahip bireylerin çok farklı belirti ve semptomları olabilir.
Belirti ve Semptomlar
L1 sendromu, farklı derecelerde şiddet derecesine sahip hidrosefali, bacakların spastisitesi ve uyarılmış başparmaklar ile karakterize hafif-şiddetli konjenital X’e bağlı bir gelişimsel bozukluktur. Sendrom, aşağıdakileri içeren bir hastalık spektrumunu temsil eder: Sylvius (HSAS) su kemeri stenozuna sahip X’e bağlı hidrosefali, MASA sendromu, X’e bağlı komplike kalıtsal spastik parapleji tip 1 ve X’e bağlı kompleks korpus kallosum agenezi.
Genetik Görülme Sıklığı
Genel olarak L1 sendromunun prevalansı bilinmemektedir; Ancak, HSAS’ın 30.000 erkekten 1’ini etkilediği tahmin edilmektedir.
Kalıtım Paterni/ Deseni
L1 sendromuna, esas olarak gelişen sinir sisteminde ifade edilen L1 hücre adezyon molekülünü kodlayan L1CAM genindeki (Xq28) mutasyonlar neden olur. Geniş klinik spektrumun açıklanmasıyla bugüne kadar 240’dan fazla farklı mutasyon bildirilmiştir. Mutasyonların yaklaşık% 7’sinin de novo olduğu rapor edilmiştir.
Bu durum X’e bağlı bir desende kalıtsaldır. Bozukluğa neden olan mutasyona uğramış gen, her bir hücrede bulunan iki cinsiyet kromozomundan biri olan X kromozomunda bulunuyorsa, X ile bağlantılı bir durum olarak kabul edilir. Sadece bir X kromozomu olan erkeklerde, her hücrede genin tek kopyasındaki bir mutasyon, duruma neden olmak için yeterlidir. X kromozomunun iki kopyasına sahip olan kadınlarda, her hücrede genin değiştirilmiş bir kopyası durumun daha az ciddi özelliklerine yol açabilir veya hiçbir belirti veya semptomlara neden olmayabilir. X’e bağlı kalıtımın bir özelliği, babaların X’e bağlı özellikleri oğullarına geçirememeleridir.
Teşhis Yöntemleri ve Tedavileri
Tanı yöntemleri:
Erkek hastalarda tanı, karakteristik klinik ve nöropatolojik bulgular ve X-bağlantılı bulaşma ile tutarlı bir aile öyküsü temelinde yapılır. Manyetik rezonans görüntüleme (MRG) veya otopsi ile tespit edilen piramitlerin iki taraflı yokluğu, pratikte, sendromun patognomonik bir özelliğidir. Teşhis L1CAM geninin moleküler genetik testi ile doğrulanabilir.
Ayırıcı tanı:
Diğer hidrosefali ve spastik parapleji bozuklukları ekarte edilmelidir. Pediatrik / nörolojik / klinik genetik çalışmaları, olası bireysel hastalıkların teşhisini sağlar.
Doğum öncesi tanı:
Bir aile üyesinde L1CAM hastalığına neden olan bir mutasyon tespit edilmişse, dişi taşıyıcılarda prenatal test yapılabilir. Koryon villus örneklemesi veya amniyosentez ile elde edilen hücrelerde kromozom analizi ile fetal cinsiyetin belirlenmesinden sonra, fetal hücreler bilinen hastalığa neden olan mutasyon için taranabilir. Kızlar etkilenebilir ve kadın fetüslerde 20 haftada ultrason önerilir. Bununla birlikte, 20 haftadaki normal fetal ultrason bozukluğu ekarte etmez: bu aşamada hidrosefali yokluğu erkek fetüsün etkilenmediğini garanti etmez.
Belirtilerin tedavisi:
L1 sendromunun tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Pediatri, çocuk nörolojisi, beyin cerrahisi, rehabilitasyon ve klinik genetik alanlarında uzmanlığa sahip çok disiplinli bir ekibi dahil etmek en iyi yöntemdir. Beyin omurilik sıvısının (BOS) şantlanması, kafa içi basıncı azaltmak için gerektiği gibi yapılmalıdır. Bazı hafif vakalarda, tendon transferi ve / veya atel başparmak fonksiyonunu iyileştirebilir. Uyarılmış başparmaklar için düzeltici cerrahi belirtilmemiştir. Zihinsel engellilik oldukça değişkendir ve gelişimsel ilerleme izlenmeli ve danışmanlık verilmelidir. Erken müdahale L1 sendromlu çocukların potansiyellerine ulaşmalarını sağlamak için önemlidir. Etkilenen çocuklara faydalı olabilecek özel hizmetler arasında özel eğitim, özel sosyal destek, fizik tedavi ve / veya diğer tıbbi, sosyal ve / veya mesleki hizmetler yer alabilir. Genetik danışmanlık, etkilenen bireylerin aile bireyleri için önerilmektedir.
Prognoz:
Hidrosefali erken bebeklik döneminde ölü doğum veya ölüme neden olabilir. Prognoz, tezahürlerin ciddiyetine bağlıdır.
Hastalığın Diğer İsimleri
- eklenmiş başparmak-zihinsel gerilik sendromu
- uyarılmış başparmak-zeka geriliği sendromu
- corpus callosum hypoplasia, mental retardation, adducted thumbs,
- korpus kallosum hipoplazisi, zihinsel gerilik, bağımlı baş parmaklar,
- spastik parapleji, hidrosefali sendromu
- CRASH sendromu
- mental retardation-clasped thumb syndrome
- mental retardasyonla büzüşen başparmak sendromu
- X’e bağlı hidrosefali sendromu
Kaynak