Genel Bilgi
Mukopolisakkaridoz tip I (MPS I), vücudun birçok bölümünü (multisistem) etkileyen nadir bir genetik hastalıktır. MPS I, en erken yaşta ortaya çıkan şiddetli formlardan çocuklukta çok belirgin hale gelmeyen daha az şiddetli formlara kadar değişen bir hastalık yelpazesi olarak düşünülür. Hurler sendromu hastalığın erken yaşlarda ortaya çıkan en şiddetli formudur. Hurler sendromu; iskelet anormallikleri, bilişsel bozukluklar, kalp rahatsızlıkları, solunum problemleri, genişlemiş karaciğer ve dalak, karekteristik surat ile karakterize edilmiştir.
Genetik Değişiklikler ve Etken Faktörler
Hurler sendromunda IDUA genindeki (4p16.3) mutasyon glikozaminoglikanların (GAGs) yıkılmasında rol oynayan alfa-L-iduronidaz enziminde eksikliğe yol açarak dermetan sülfat ve heperan sülfatın lizozomal birikimine yol açar.
Belirti ve Semptomlar
Şiddetli MPS I yani Hurler sendromu, çoklu organ ve dokuları içeren kronik ve progresif bir hastalık seyri ile karakterizedir .
Hurler sendromlu çocuklar doğumda normal görünür Ancak kasık veya göbek fıtıkları olabilir. Karakteristik görünüm yaşamın ilk yıllarında gelişir. Şiddetli MPS I için ortalama tanı yaşı yaklaşık dokuz aydır.
Yüz özelliklerinin 3-6 aylıkken hafifçe kabalaşması genellikle tespit edilen ilk anormalliktir. Kafa büyük ön kemikler yüzünden şişkindir.
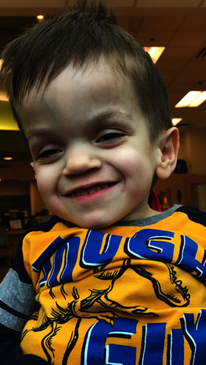
Figür: Mukopolisakkaridoz tip I olan bir çocuk
Kaynak: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i#definition
Kalp kapağı anormallikleri, beyinde sıvı birikmesi, genişlemiş bir karaciğer ve dalak (hepatosplenomegali) ve geniş bir dil (makroglossia) görülür. Bazı vakalarda sık sık üst solunum yolları enfeksiyonları ve uyku apnesi görülebilir.
Progresif hepatosplenomegalinin neden olduğu karın şişkinliği yaygındır. Organ büyümesi görülse de, karaciğerde ve dalakta glikozaminoglikanların depolanması organ işlev bozukluğuna yol açmaz.
Kornea bulanıklığı yaygındır. Hastalık önemli görme kaybına neden olabilir. Etkilenen kişilerde işitme kaybı ve tekrarlayan kulak enfeksiyonları da olabilir.
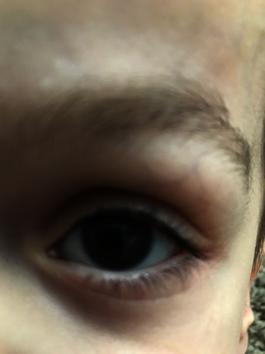
Figür: Mukopolisakkaridoz tip I ile ilişkili kornea bulanıklığı
Kaynak: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i#definition
Üç yaşına gelince doğrusal büyüme azalır. Omurga gövdelerinin kusurlu ossifikasyon merkezleri, düzleşmiş ve gagalanmış omurlara ve sonrasında da omurga deformitesine yol açar. Bu bazı vakalarda hareketliliği engeller. Çoklu iskelet anormallikleri anlamına gelen distostoz multipleksler görülür.
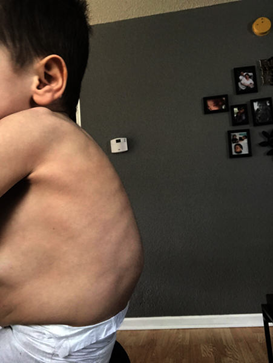
Figür: Mukopolisakkaridoz tip I ile ilişkili kavisli omurga
Kaynak: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i#definition
Karpal tünel sendromu bu bozukluğu olan birçok çocukta gelişir ve el ve parmaklarda uyuşma, karıncalanma ve güçsüzlük ile karakterize edilir.
Erken psikomotor gelişim normal olsa da, gelişimsel gecikme 18 aylıkken genellikle belirgindir. Entelektüel kapasitede ölçülebilir bir azalma bundan sonra aylık olarak gerçekleşir. İlerleyen dönemlerde zihinsel yeteneklerde yavaş bir düşüş gerçekleşir. Sekiz ila on yaşları arasındaki ölüm zamanlarında, çoğu çocuk zihinsel olarak engellidir.
Genetik Görülme Sıklığı
MPS1’in Hurler alt türünün yaygınlığının Avrupa’da 1 / 200.000 olduğu tahmin edilmektedir.
Hastalıkla İlişkili Genler
Hurler sendromu; 4p16 kromozomu üzerindeki alfa-L-iduronidaz kodlayan IDUA geninde homozigoz veya bileşik heterozigoz mutasyondan kaynaklanır.
Kalıtım Paterni
Hurler sendromu otozomal resesif kalıtılan genetik bir hastalıktır. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve semptomlarını göstermezler.
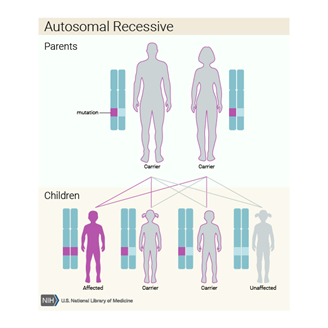
Figür: Otozomal resesif kalıtım paterni
Kaynak: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i#inheritance
Teşhis Yöntemleri ve Tedaviler
Klinik bulgular
- Kaba yüz özellikleri,
- Orta kulak iltihabı dahil erken sık görülen üst solunum yolu enfeksiyonları,
- Kasık veya göbek fıtığı,
- Hepatosplenomegali,
- Karakteristik iskelet ve eklem bulguları (gibbus deformitesi; eklem hareket açıklığının sınırlandırılması),
- Karakteristik oküler bulgular (kornea bulanıklığı)
Klinik bulgular hastalık şiddetine göre değişir. Ancak sadece klinik bulgular hastalık tanısı için yeterli değildir. Bu yüzden laboratuvar analizleri yapılır.
Laboratuvar analizleri, idrarda artan glikozaminoglikanlarının yani heparan ve dermatan sülfatın kalitatif (GAG elektroforezi) ve kantitatif (toplam idrar ürronik asit ölçümü) analizine dayanır. Ancak Ne nicel ne de nitel yöntem, MPS I de dahil olmak üzere spesifik bir lizozomal enzim eksikliğini teşhis edemez; ancak, yöntemlerden biri veya her ikisi ile tespit edilen bir anormallik, bir MPS bozukluğunun muhtemel varlığını gösterir.
Kesin tanı için moleküler genetik testler yapılabilir. Moleküler test yaklaşımları; IDUA geninde Gen hedefli silme/ çoğaltma yapılmasına ya da Dizi analizine dayanır.
Son bir teşhis yöntemi olarak da Alfa-L-iduronidaz enzim aktivitesi çoğu dokuda ölçülebilir; tipik olarak periferik kan lökositleri, plazma veya kültürlenmiş fibroblastlar kullanılır. Neredeyse tüm MPS’li bireylerde, teşhis için kullanılan standart numunelerde tespit edilebilir bir enzime rastlanaz.
Mukopolisakkaridoz tip I tanısı, karakteristik semptomların tanımlanması, ayrıntılı bir hasta ve aile öyküsü, ayrıntılı bir klinik değerlendirme ve çeşitli uzmanlık testlerine dayanır. Karakteristik erken belirtileri olan bebeklerde tanıdan şüphelenilebilir. Halen MPS I için yenidoğan taraması birçok alanda uygulanmaktadır.
Tedavi
1. Eksik enzimin değiştirilmesi: Lizozomal enzimler, hücreler tarafından alınabildikleri ve kullanılabildiklerinden dolayı eşsiz proteinlerdir. Bu nedenle, MPS I’yi tedavi etmenin potansiyel bir yolu, hastalara eksik aldıkları alfa-L-iduronidaz enzimini vermektir. Bu 2 şekilde yapılabilir.
a. Bunlardan biri, enzim replasman tedavisi (ERT) olarak da bilinen saflaştırılmış enzim ile beslemektir. Enzim replasman tedavisi, eksik enzimin alfa-L-iduronidazın genetik olarak tasarlanmış (rekombinant) bir formla değiştirilmesini içerir. Tüm Hurler hastalarına ERT önerilmektedir ve nörolojik olmayan semptomları hafifleten yaşam boyu bir terapidir. ERT’nin erken kullanımının, bu durumun bazı klinik özelliklerinin gelişimini geciktirdiği veya hatta engellediği gösterilmiştir. Bu, enzimi haftada bir intravenöz yoluyla bir hastaya infüze etmek suretiyle gerçekleştirilir.
b. Hematopoetik kök hücre nakli (HSCT), hayatta kalma süresini uzatabildiği, nörobilişimi koruyabildiği ve bazı somatik özellikleri iyileştirebileceği için, 2.5 yaşın altındaki (ve bu yaş sınırının üzerindeki seçilmiş hastalarda) Hurler sendromlu hastalar için tercih edilen tedavi yöntemidir.HSCT, hastalığın seyrinde, gelişimsel bozulma başlamadan önce erken yapılmalıdır. HSCT sağkalımı artırabilir, büyümeyi artırabilir, yüz kalınlığını ve hepatosplenomegaliyi azaltabilir, işitmeyi iyileştirebilir ve kalp ve solunum semptomatolojisinin doğal geçmişini değiştirebilir. HSCT’nin iskelet ve eklem belirtileri veya kornea bulanıklığı üzerinde daha az etkisi vardır. HSCT, transplantasyon sırasında hafif fakat anlamlı olmayan bilişsel bozulma olan çocuklarda bilişsel gerileme sürecini yavaşlatabilir. HSCT ile ilişkili morbidite ve mortalite nedeniyle, günümüzde öncelikle ağır MPS I olan çocuklar için önerilmektedir.
2. Hastalığın spesifik semptomlarını hafifletmek: Mukopolisakkaridoz tip I tedavisinin önemli bir bileşeni, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Bu, uzmanlardan oluşan bir ekibin koordine çabalarını gerektirir
3. Etkilenen bireyler ve aileleri için genetik danışmanlık önerilir. Tüm aile için psikososyal destek de şarttır.
Hastalığın Diğer İsimleri
- Hurler hastalığı
- MPS1H
- MPSIH
- Mukopolisakkaridoz tip 1H
Kaynakça
- https://rarediseases.org/rare-diseases/mucopolysaccharidosis-type-i/ /
- https://www.omim.org/entry/607014?search=hurler%20syndrome&highlight=%28syndrome%7Csyndromic%29
- https://rarediseases.info.nih.gov/diseases/12559/hurler-syndrome
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=12381&Disease_Disease_Search_diseaseGroup=Hurler-syndrome&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Hurler-syndrome&title=Hurler%20syndrome&search=Disease_Search_Simple
- https://www.ncbi.nlm.nih.gov/books/NBK1162/
- https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i#definition