Genel Bilgi
Pitt-Hopkins sendromu zihinsel engellilik, gelişimsel gecikme, solunum problemleri, nöbetler (epilepsi), tipik yüz yapısı ve yüksek miyopi özellikleri ile karakterize olan genetik bir sendromdur. TCF4 geninin mutasyonu sonucu ortaya çıkar ve otozomal dominant karakterdedir. İlk defa 1978 yılında David Pitt ve Ian Hopkins tarafından tanımlanmıştır. Nedeni bilinse de tedavisi için bir ilacı bulunmamaktadır.
Genetik Değişiklikler/Etken Faktörler
Pitt-Hopkins sendromuna TCF4 genindeki mutasyon ya da 18. kromozomun TCF4 genini içeren bölgesindeki delesyon sebep olmaktadır. Otozomal baskın (dominant) olarak kalıtılır fakat ailesinde Pitt-Hopkins sendromu öyküsü olmadığı halde de mutasyon sonucu ortaya çıkmaktadır.
TCF4 geni Pitt-Hopkins sendromundan başka Şizofreni, Otizm, Fuchs Kornea Distrofisi ve Karaciğer hastalığı olmak üzere çeşitli hastalıklarda da rol oynar.
Belirti ve Semptomlar
Pitt-Hopkins sendromunun birçok belirti ve semptomu bulunmaktadır. Semptomlar ve şiddetleri kişiden kişiye değişebilir. Erken semptomlar bebek doğduktan sonra bir sene içinde görülmektedir. Çok düşük kas tonusu (hipotoni) ve gelişim geriliği görülmektedir. Bazı bebeklerde baş boyutu küçüktür (mikrosefali). Çocuklar yürümeye beklenenden aylar hatta yıllar sonra başlayabilir. Bazıları ise bağımsız olarak yürüme yeteneğini kazanamazlar. Konuşma gecikmiştir. Bazıları birkaç kelime söylemeyi öğrenebilirken çoğu konuşamaz. Bununla birlikte bazıları basit yönergeleri anlayıp uygulayabilir. Zihinsel engellilik seviyesi genellikle orta ila şiddetlidir.
Yüz özellikleri çukur gözler, çıkık burun, aşağı yönelmiş burun ucu, geniş burun delikleri ve burun kemeri, kısa filtrum, geniş ağız, geniş aralıklı dişler ve çıkık çene ile ayırt edilir. Bu özellikler yaş ile daha belirgin hale gelir. (Şekil 1)
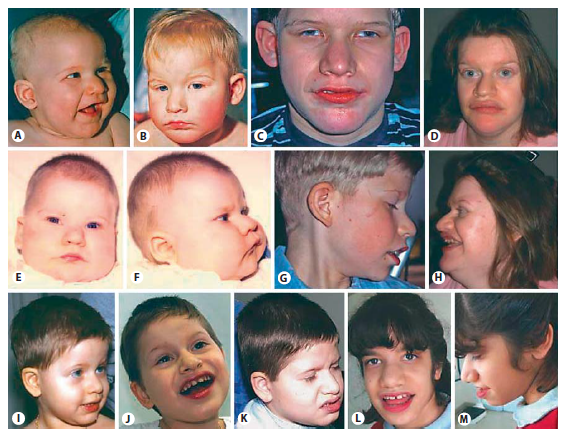
Şekil 1. Pitt-Hopkins sendromu hastalarında yüz özellikleri. Hasta 1 A (6 aylık), B (18 aylık) ve C (14 yaş). Hasta 6 D ve H (29 yaş). Hasta 2 E, F (6 aylık) ve G (11 yaş). Hasta 3 I ( 3 yaş), J (6 yaş) ve K (8 yaş). Hasta 4 L ve M (12.5 yaş).
Otizm spektrum bozukluğu belirtileri görülebilir. Beslenme sırasında alışılmadık davranışlar, agresif davranışlar, anksiyete, stereotipik el ve kafa hareketleri görülür. Çoğu çocukta mutlu bir yüz ifadesi vardır. Uykuda kaybolan, soluk alıp verme problemi görülür. Bunlar genelde anksiyete, heyecanlanma ya da yorulma sonucu gerçekleşir. Hızlı nefes alıp verme ardından nefes almamaya veya nefes almaya çalışma davranışları birbirini takip eder. Pitt-Hopkins sendromuna sahip kişilerin neredeyse yarısında nöbet görülür. Nöbetler çocukluk ya da gençlik döneminde herhangi bir zamanda başlayabilir. Uyuya kalma ya da uykusuzluk gibi sorunlar görülebilir. Hastaların yarısından azında gastroözofageal reflu görülür. Miyopi iki yaşından önce görülebilir ve şiddetli olabilir. Bunun haricinde şaşılık ve astigmatizm de görülür. İskelet sisteminde skolyoz, düztabanlık, yumru ayak, küçük el ve ayaklar, kavisli ya da bükük parmaklar, geniş parmak uçları ya da gittikçe incelen parmaklar görülebildiği rapor edilmiştir. Ayak parmakları üst üste binmiş halde olabilir. Acı eşikleri yüksektir. Etkilenen erkeklerin yaklaşık üçte birinde testislerden biri ya da her ikisi de skrotuma inmemiştir.
Genetik Görülme Sıklığı
Pitt-Hopkins sendromunun oldukça nadir olduğu düşünülmektedir. Dünya çapında 500 kişinin bu sendroma sahip olduğu rapor edilmiştir. Kadın ve erkekte görülme sıklığı eşittir.
Kalıtım Paterni/Deseni
Otozomal baskın (dominant) olarak kalıtılır. Baskın genetik hastalıkların ortaya çıkması için anormal genden bir tane bulunması yeterlidir. Bu gen anne veya babadan gelmiş olabilir.
Üreme hattı mozaikliği görülebilir. Bu durumda anne veya baba TCF4 gen mutasyonunu vücut hücrelerinde taşımaz yani etkilenmemiştir. Fakat üreme hücreleri (yumurta ya da sperm) mutasyona sahiptir. Yumurta veya spermlerden biri mutasyona sahipse hastalık yavru bireyde görülür. Ebeveynlerin kanlarından yapılan testte mutasyon negatif çıksa bile sonraki çocuklarının da etkilenmiş olma olasılıkları yaklaşık %1-2 kadardır.
Teşhis Yöntemleri ve Tedaviler
Pitt-Hopkins sendromunun teşhisi detaylı hasta hikayesi, klinik bulgular ve semptomun karakteristik özelliklerinin tanımlanmasıyla gerçekleştirilir. Ancak bu sendrom ve diğer bazı nörolojik bozukluklarda görülen semptomlar benzerlik göstermektedir. EEG ve beyin MRI görüntülemesiyle ve genetik analizler sonucunda TCF4 geninde meydana gelmiş mutasyon ya da delesyon tespit edilerek teşhis desteklenir.
Bilinen bir tedavisi yoktur. Tedaviler semptomları gidermek için uygulanır.
Hastalıkla İlişkili Genler
TCF4 genindeki mutasyonlar Pitt-Hopkins sendromuna neden olmaktadır. Bu gen 18.kromozomun uzun kolunda bulunmaktadır (18q21.2). TCF4 geni, vücutta birçok fonksiyonda kritik rol oynayan proteinlerin yapımı için bilgiler içerir. Bu gende mutasyon gerçekleştiğinde oluşan protein hatalı ve etkisiz olabilir, eksik veya fazla üretilebilir. TCF4 geni, transkripsiyon faktörü olarak da bilinen TCF4 proteinini sentezlenmesini sağlar. Bu proteinin farklı gelişim evrelerinde hücre farklılaşması ve hücre ölümü gerçekleştirmek gibi önemli görevleri vardır. İnsan gelişiminin erken dönemlerinde sinir sistemi gelişimiyle ilişkili olarak yüksek düzeyde eksprese edilir.
Hastalığın Diğer İsimleri
Hastalık PHS ve PTHS kısaltmalarıyla da adlandırılır.
Kaynaklar
Peippo M., Ignatius J. Pitt-Hopkins Syndrome. Mol Syndromol 2011;2:171–180
Sweatt J. D. Pitt–Hopkins Syndrome: intellectual disability due to loss of TCF4-regulated gene transcription. Experimental & Molecular Medicine (2013) 45