Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Pallister-Hall sendromu (PHS), vücudun birçok bölümünün gelişimini etkileyen genetik bir hastalıktır. Ortak özellikler arasında ekstra parmaklar ve / veya ayak parmakları (polidaktil), parmaklar veya ayak parmakları arasında ekstra cilt (sindaktil), beyinde hipotalamik hamartom denilen bir anormal büyüme ve iki üçlü epiglotis olarak bilinen hava yolunun bir malformasyonu bulunur. Nadir durumlarda bifid epiglot, solunum yetmezliğine yol açabilir. Çoğu durumda hipotalamik hamartom sorun yaratmasa da, bazı durumlarda nöbetler, büyüme hormonu eksikliği, erken ergenlik veya kortizol eksikliğine neden olabilecek birçok hormonun (panhipopitüitarizm) eksikliği gibi nörolojik sorunlara neden olabilir. PHS’nin diğer semptomları arasında deliksiz anüs, böbrek anomalileri, kalp defektleri, küçük genital organlar, parmak eksikliği, tırnak problemleri, yarık damak, bifid uvula ve gelişim gecikmesi ve davranış problemleri sayılabilir. GLI3 genindeki mutasyonlar Pallister-Hall sendromuna neden olur. Bu gen, genlerin belirli hücrelerde açılıp kapatılmayacağını düzenleyen bir işlem olan gen ekspresyonunu kontrol eden bir protein yapmak için talimatlar sağlar. Gelişim sırasında belirli zamanlarda belirli genlerle etkileşime girerek, GLI3 proteini doğumdan önce birçok organ ve dokunun normal şekillenmesinde (desenlenmesinde) rol oynar.
Pallister-Hall sendromuna neden olan mutasyonlar tipik olarak GLI3 proteininin anormal derecede kısa bir versiyonunun üretilmesine yol açar. Hedef genleri açıp kapatabilen normal GLI3 proteininin aksine, kısa protein sadece hedef genleri kapatabilir (bastırabilir). Araştırmacılar, protein işlevindeki bu değişikliğin erken gelişimi nasıl etkilediğini belirlemek için çalışmaktadır. GLI3 mutasyonlarının polidaktil, hipotalamik hamartom ve Pallister-Hall sendromunun diğer özelliklerine neden olabileceği kesin değildir.
Belirti ve Semptomlar
PHS’li çoğu hasta doğumda üçüncü veya dördüncü dereceden iskelet polidaktili veya postaksiyal polidaktili görülürken, beraberinde her ikisine de kütanöz sindaktili ve tırnak displazisi eşlik edebilir. Yüz özellikleri kısa burun, düz burun köprüsü ve alçak ayarlanmış, arka acılı kulakları içerebilir. Bazı hastalarda yarık damak, yarık uvula ve multipl bukal frenula bildirilmiştir. Asemptomatik bir bifid epiglotis neredeyse patognomoniktir, ancak bazı hastalarda potansiyel olarak ölümcül solunum yetmezliğine yol açan daha ciddi posterior laringeal yarıklar görülür. Hipotalamik hamartom genellikle asemptomatiktir, ancak panhipopituitarizm ile ilişkili olabilir. Akut primer adrenal yetmezlik ciddi vakaların yani sıra, daha hafif adrenal yetmezlik formlarında da görülebilir. Erken ergenlik bazı durumlarda kendini gösterir. Nörolojik tutulum, gelastik epilepsiyi (yüz ekşitmeden, gülüşmekten veya kahkaha olarak ortaya çıkan nöbetler) veya diğer nöbet tiplerini içerebilir. Vajinal atrezi veya hidrometrokolpos, microphallus veya kriptorşidizm de dahil olmak üzere böbrek agenezisi veya displazisi ile diğer genitoüriner anomaliler bildirilmiştir. Diğer bulgular arasında intrauterin gelişme geriliği, anormal akciğer lobasyonu, mezomelik kısalması ile genelleşmiş iskelet displazisi ve uzuvların radyal yaylanması, imperforat anüs ve konjenital kalp defektleri sayılabilir.
Genetik Görülme Sıklığı
Bu durum çok nadirdir; prevalansı bilinmemektedir. Bugüne kadar yaklaşık 100 hasta bildirilmiştir.
Kalıtım Paterni/Deseni
Bu durum otozomal dominant paternde kalıtsaldır, bu da her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir. Bazı durumlarda, etkilenen bir kişi, etkilenen bir ebeveynden GLI3 geninde bir mutasyon devralır. Diğer vakalar, gendeki yeni mutasyonlardan kaynaklanır ve ailelerinde hastalık öyküsü olmayan kişilerde ortaya çıkar.
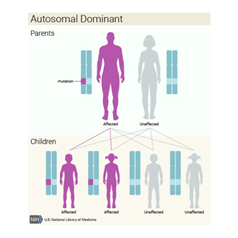
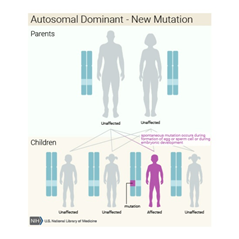
Kalıtım paterni : https://ghr.nlm.nih.gov/condition/pallister-hall-syndrome#inheritance
Teşhis Yöntemleri ve Tedaviler
Iafolla ve diğerleri (1989) manyetik rezonans görüntülemenin en değerli tanı aracı olduğuna işaret etmişlerdir; BT taramasının tümörü kaçırdığı bildirildi.
Pallister-Hall sendromu üzerine uluslararası bir atölye çalışması (Biesecker ve diğerleri, 1996) bu varlık için minimal tanı kriterleri geliştirmiştir. Bir ailedeki indeks olgusunda, tanı kriterlerini karşılamak için hem hipotalamik hamartom hem de merkezi polidaktili olmalıdır. İndeks olgunun birinci derece akrabalarında hipotalamik hamartom veya polidaktili (merkezi veya postaksiyal) bulunmalı ve otozomal dominant paternde veya gonadal mozaikliği ile uyumlu bir şekilde kalıtım gösterilmelidir. Şüpheli olguların klinik değerlendirmesi için öneriler sunuldu. Biesecker ve diğerleri (1996), hipotalamik hamartomun PHS’ye özgü olmadığı sonucuna varmıştır.
Hastalıkla İlişkili Genler
Pallister-Hall sendromuna GLI3 genindeki mutasyonlar neden olur. Kalıtım otozomal dominanttır, ancak vakaların yaklaşık dörtte birinde Pallister Hall sendromu yeni (de novo) bir mutasyondan kaynaklanır
Hastalığın Diğer İsimleri
- Hall-Pallister sendromu
- PHS
Referanslar
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2130&Disease_Disease_Search_diseaseGroup=pallister-hall-syndrome&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Pallister-Hall-syndrome&title=Pallister-Hall%20syndrome&search=Disease_Search_Simple
- https://ghr.nlm.nih.gov/condition/pallister-hall-syndrome#genes
- https://www.omim.org/entry/146510?search=Pallister%20Hall%20Syndrome&highlight=%28syndrome%7Csyndromic%29%20hall%20pallister
- https://rarediseases.org/gard-rare-disease/7305/pallister-hall-syndrome/