Genel Bilgi
Hiper-Ig E Sendromu bağışıklık sisteminde yetmezliğin yol açtığı semptomlarla seyreden nadir görülen bir hastalıktır. Anne-babadan otozomal baskın ya da otozomal çekinik olarak aktarılabilir veya hastalıklı bireyde yeni bir mutasyonun oluşmasıyla ortaya çıkabilir.
Bu hastalığın otozomal baskın kalıtımıyla ortaya çıkan formu olan Otozomal Baskın Hiper-IgE Sendromu (OB-HIES) diğer bir adıyla Job Sendromu, vücutta başlıca bağışıklık sistemini etkilemektedir. Bu hastalarda tekrarlayan enfeksiyonlar (zatürre, cilt enfeksiyonları), egzama, kemik ve diş anormallikleri görülür. Hastaların kanında İmmunglobulin E (IgE) antikoru yüksektir. (1)
Genetik Değişiklikler/Etken Faktörler
Vakaların çoğunda STAT3 genindeki mutasyon hastalığa sebep olmaktadır. Bu genin kodladığı STAT3 proteini, başta T hücreleri olmak üzere bağışıklık sistemindeki hücrelerin olgunlaşmasında rol oynayan bir proteindir. Bağışıklık sistemi hücreleri normalde bakteri ve mantar gibi hastalık yapıcı etkenlere karşı vücut savunmasını sağlar.
STAT3 genindeki mutasyonlar STAT3 proteininin yapı ve fonksiyonlarında değişikliklere yol açar. Sonuç olarak bağışıklık sisteminde anormallikler oluşur ve bu da vücudu, özellikle de akciğer ve cilt dokusunu, bakteri ve mantar enfeksiyonlarına duyarlı hale getirir. STAT3 proteini ayrıca kemik dokusundaki yapım ve yıkım olaylarına da katılır, bu yüzden kemik ve dişlerde anormallikler görülmektedir. STAT3 gen mutasyonunun IgE seviyelerini hangi mekanizmayla yükselttiği bilinmemektedir. (2)
Belirti ve Semptomlar
OB-HIES yenidoğan döneminde döküntülerle ortaya çıkar ve bağışıklık sistemini, kemikleri, bağ dokusu ve diş gelişimini etkiler. Ciltte egzama ve tekrarlayan enfeksiyonlar görülür, bu enfeksiyonlar içi iltihap dolu apseler oluşturur. Tekrarlayan zatürre gibi akciğer enfeksiyonları akciğerde içi hava dolu keselerin oluşmasına neden olur (Pnömatosel). Tekrarlayan solunum yolu enfeksiyonları kronik solunum yetmezliğine yol açabilir. Bu hastalarda Candida (mantar) enfeksiyonları da sık görülmektedir. Hastalarda tipik bir yüz görünümü vardır: Pürüzlü cilt, yüzde asimetri, alın ve çenede çıkıklık, gözlerde çukurluk, geniş burun köprüsü ve dolgun burun ucu. Hastalarda tekrarlayan kemik kırıkları ve skolyoz görülebilir; diş yapımında bozukluklar vardır. Anevrizma, tromboz ve PDA gibi damarsal bulgular da bildirilmiştir. Gözde ksantelezma, arpacık, göz kapağı nodülleri, şaşılık ve retina dekolmanı gibi komplikasyonlar olabilir. Hastalarda otoimmün ve lenfoproliferatif hastalık görülme riski artmıştır. (3)
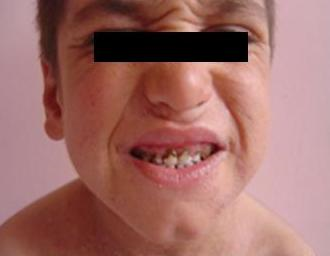
Görsel 1: OB-HIES tipik yüz görüntüsü (4)
Genetik Görülme Sıklığı
OD-HIES nadir bir hastalıktır ve görülme sıklığı 1/1.000.000’dur. (3)
Kalıtım Paterni
Bu hastalık otozomal baskın kalıtılır, yani mutasyonlu geni taşıyan herkes hastalığa sahiptir. Hastaların yaklaşık yarısında mutasyonlu gen, etkilenmiş anne ya da babadan kalıtılır yani tek bir ebeveynin hastalıklı geni çocuğa aktarması, hastalığın oluşumu için yeterlidir. Diğer vakalarda ise anne ya da babada hastalık yoktur, hastalığın sorumlusu gende oluşan yeni mutasyonlardır. (2)
Teşhis Yöntemleri ve Tedaviler
Tanıda klinik bulgularla beraber laboratuvar bulguları değerlendirilir. Kanda 2000 U/ml’nin üstündeki IgE değerleri hastalığın tanısı için anlamlıdır, hatta çoğu zaman IgE değerleri 5000 U/ml’den de yüksek görülür. STAT3 mutasyonunun genetik olarak gösterilmesi tanıyı doğrular.
Bu hastalığın kesin bir tedavisi yoktur, tedavi yaklaşımı enfeksiyonların önlenmesine yöneliktir, bu amaçla uzun dönemli antibiyotik ve antifungaller (mantar ilacı) kullanılır. OB-HIES yaşam boyu süren kronik bir hastalıktır ve oluşan her yeni enfeksiyon tedavi edilmelidir. Akciğer apseleri için cerrahi müdahale gerekebilir. (3) Egzama için nemlendiriciler ve kullanımı sınırlı tutulmakla birlikte topikal steroidler kullanılabilir. Apselerin tedavisi drenajdır (apsenin boşaltılması). Kemik iliği nakli Otozomal Resesif Hiper IgE Sendromu için tedavi edici olsa da, OB-HIES tedavisinde önerilmemektedir. (5)
Hastalıkla İlişkili Genler
17. kromozomun uzun kolunda lokalize olan STAT3 geni vakaların büyük bir kısmından sorumludur. STAT3 gen mutasyonu olmayan kişilerde hastalığın genetik nedeni bilinmemektedir. (2)
Hastalığın Diğer İsimleri
- Job Sendromu
- AD-HIES
- Otozomal Dominant Hiper-IgE Sendromu
- Buckley sendromu
- STAT3 yetmezliği
Kaynaklar
1)https://rarediseases.info.nih.gov/diseases/6800/hyper-ig-e-syndrome-autosomal-dominant
2)https://ghr.nlm.nih.gov/condition/autosomal-dominant-hyper-ige-syndrome#
3)https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=2314&lng=EN
4 https://step1.medbullets.com/immunology/105008/hyper-ige-syndrome–job-syndrome
5)https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/hyper-ige-syndrome