Genel Bilgi
Meckel sendromu, böbreklerde çoklu kistler, kafatasındaki bir açıklıktan (oksipital ensefalosel) beynin bir kısmının çıkıntısı ve ekstra parmaklar (polidaktili) ile karakterize çok ciddi bir hastalıktır. Etkilenen çocuklarda baş ve yüz, karaciğer, akciğerler, cinsel organlar ve idrar yollarını etkileyen anormallikler de olabilir. Bu ciddi sağlık sorunları nedeniyle, Meckel sendromlu bebeklerin çoğu doğumdan sonra uzun süre hayatta kalmaz. Meckel sendromu, sekiz genden birinde mutasyonlardan kaynaklanır ve otozomal-resesif bir şekilde kalıtsaldır.
Belirti ve Semptomlar
Meckel sendromu ile ilişkili spesifik semptomlar bir kişiden diğerine büyük ölçüde değişir. Etkilenen çocuklar aşağıda ayrıntılı semptomların hepsine sahip olmayacaktır. Merkezi sinir sistemi, pulmoner veya böbrek anormallikleri her zaman perinatal ölümle sonuçlanır.
Meckel sendromuyla ilişkili en yaygın merkezi sinir sistemi anormalliği, bir bebeğin kafatasında bir boşlukla doğduğu bir durum olan oksipital ensefaloseldir.
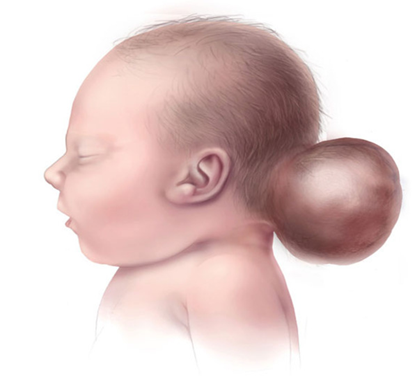
Etkilenen bebekler anormal derecede küçük bir çene (mikrognati), genişlemiş ve kusurlu kulaklar, yarık damak, yarık dudak, eğimli alın ve kısa boyun gibi farklı özelliklere sahip olabilirler. Etkilenen çocuklarda anormal derecede küçük gözler (mikroftalmi) ve gözlerin sinirlerinin az gelişmesi (optik sinir hipoplazisi veya koloboma) dahil olmak üzere göz (oküler) anormallikleri görülebilir. Böbreklerdeki çoklu kistler (multikistik böbrek displazisi) Meckel sendromuyla ilişkili en yaygın semptomdur.
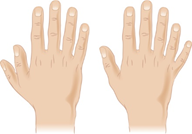
Etkilenen bireyler ayrıca ekstra parmak ve ayak parmaklarına, çoğunlukla ellerin baş parmağında ekstra parmaklara (postaksiyal polidaktili) sahip olabilirler.
Genetik Görülme Sıklığı
Yaygınlık Avrupa’da 50,000’de 1 doğum olarak tahmin edilmektedir. Canlı doğum yaygınlığının dünya çapında 13,250’de 1 ile 140,000’de 1 arasında olduğu bildirilmektedir. Canlı doğum yaygınlığı, Fin popülasyonunda (1/9,000), Belçika ve Kuveyt Bedevi popülasyonlarında (1/3,500) ve Gujarati Kızılderililerinde (1/1,300) önemli ölçüde daha yüksektir. Cinsiyet veya etnik tercih bildirilmemiştir.
Kalıtım Paterni
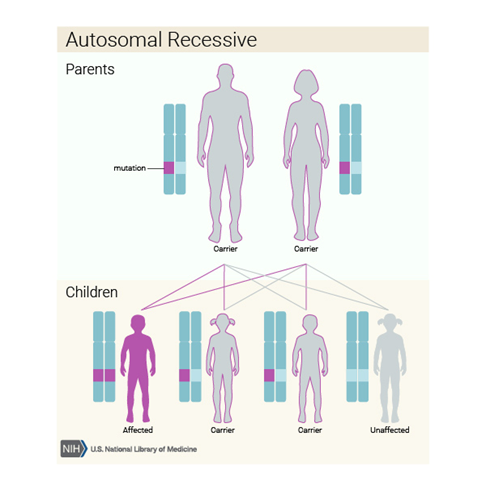
Bu durum otozomal resesif bir desende kalıtsaldır, bu da her hücredeki genin her iki kopyasının mutasyonları olduğu anlamına gelir. Otozomal resesif bir duruma sahip bir bireyin ebeveynleri her biri mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirti ve semptomlarını göstermezler.
Nüks riski %25’dir.
Nedenleri
Meckel sendromu, on üç gendeki değişikliklerden (mutasyonlar) kaynaklanabilir: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRİP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, tmem231 ve TMEM237. Bu 13 gendeki mutasyonlar tüm vakaların yüzde 75’ini oluşturur; kalan yüzde 25’in bilinmeyen genetik nedenleri vardır. Bu genlerin çoğu, Joubert sendromu adı verilen nörolojik bir bozukluktan da sorumludur ve Meckel sendromunun Joubert sendromunun aşırı öldürücü formu olduğu kavramına yol açar.
Teşhis Yöntemleri ve Tedaviler
Oksipital ensefalosel ve displastik böbrekleri gösteren fetal ultrasonografi ile tanı konabilir. MKS tanısı için üç ana malformasyondan ikisi veya bir klasik özellik ile birlikte diğer iki anomali yeterlidir. Otopsi de gerekebilir. Bozukluk genellikle 14. gebelik haftasından önce tespit edilir. Moleküler genetik testler, genetik danışmanlığa rehberlik etmek için tanıyı doğrulamak için kullanılabilir.
Ölümcül bir sonucu olan Meckel sendromu için şu anda tedavi mevcut değildir.
Hastalığın Diğer İsimleri
- Dysencephalia splanchnocystica
- Meckel-Gruber sendromu
- MKS
Referanslar