Genel Bilgi
Osteopetrozis, kemiklerin anormal şekilde yoğunlaşması ve kırılmaya yatkın olmasına neden olan nadir, kalıtsal bir iskelet hastalığıdır. Semptomlar ve şiddetleri çok değişken olabilir. Yenidoğanda ortaya çıkan, hayati tehlikesi olan belirtileri (kemik iliği yetersizliği vb.) olabildiği gibi, tesadüfi olarak röntgende bulunarak da teşhis konabilir. Semptomların şiddetine ve ortaya çıkma yaşına bağlı olarak bireyde kemik kırıkları, kısa boy, kompresif nöropati (sinirlerde baskı), tetanik kasılmaya eşlik eden hipokalsemi ve yaşamı tehdit eden pansitopeni (kan hücrelerinin eksikliği) görülebilir. Nadir vakalarda nörolojik bozulma ya da diğer vücut sistemlerinde de sorunlar görülebilir. Osteopetrozisin otozomal dominant (baskın), otozomal resesif (çekinik) veya X’e bağlı olarak kalıtılan tipleri vardır. Otozomal Dominant Osteopetrozis (Albers Schöngen Hastalığı), genelde hastalığın en hafif tipidir. Etkilenen bireylerin bazılarında hiç belirti görülmez. Belirti gösteren hastalarda ise kemik kırıkları, omurga eğriliği (skolyoz) ya da diğer omurga anomalileri, kalçada eklem iltihabı (artrit) ve kemik iltihabı (osteomiyelit) bulunabilir. Bu problemler genelde çocukluk çağının sonlarında ya da ergenlikte belli olur. Tedavi yaklaşımları semptomlara ve belirtilerin şiddetine göre değişiklik gösterir.
Belirti ve Semptomlar
Belirtiler kişiden kişiye birçok farklılık gösterebilir. Başlıca belirtiler şunlardır:
- Kemiklerde şekil anomalileri
- Kemiği besleyen damarlarda kan akımının azalması sonucu doku ölümü (avasküler nekroz)
- Kemik ağrıları
- Yüz felci (Bell paralizi)
- Eklem çıkıkları
- Kafatasının normalden büyük olması (makrosefali)
Görülme Sıklığı
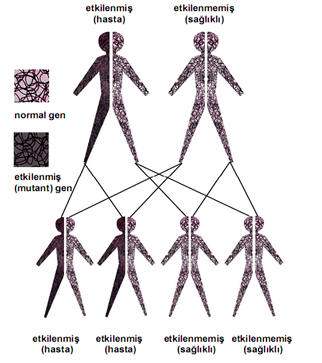
Albers Schöngen Hastalığı, osteopetrozis tipleri arasında en sık görülenidir. 20,000 kişiden 1’ini etkiler. Otozomal dominant kalıtım gösterdiği için bireyde genin bir anormal kopyasının bulunması hastalığın oluşması için yeterlidir. Genelde mutasyonlu gen, hastalığa sahip ebeveynden aktarılır.
Kaynak: http://www.eurogentest.org/index.php?id=268
Genetik Değişiklikler
En az 9 gende meydana gelen mutasyonların, farklı tiplerdeki osteopetrozise neden olabildiği bilinmektedir. CLCN7 genindeki mutasyonlar, Otozomal Dominant Osteopetrozis vakalarının yaklaşık %75’inden sorumludur. Diğer genlerdeki mutasyonlar daha az yaygındır. Osteopetrozis vakalarının %30’unda hastalığın nedeni bilinmemektedir.
Osteopetrozisle ilişkili genler, osteoklast denen özelleşmiş hücrelerin yapımından, gelişiminden ve fonksiyonunun düzenlenmesinden sorumludurlar. Osteoklast hücreleri, kemik yapım-yıkım döngüsünde (remodelasyon) kemik dokusunun yıkımından sorumludur. Bu, eski kemik dokusunun yıkılıp onun yerine yeni kemik oluşumunun gerçekleştiği normal bir döngüdür. Kemik remodelasyonu, kemiğin sağlıklı ve güçlü kalmasını sağlamak amacıyla çok sıkı bir şekilde kontrol edilir. Osteopetrozise neden olan genlerdeki mutasyonlar, osteoklast hücrelerinin kaybına veya anormal hücrelerin oluşumuna yol açar. Düzgün işlev göremeyen osteoklastlar, yeni kemik oluşurken eski kemiğin yıkımını sağlayamaz. Sonuç olarak kemikler yoğun/ kalın bir hal alır. Ayrıca yapıları da bozuk olan kemikler kırılmaya yatkın olurlar.
Teşhis Yöntemleri
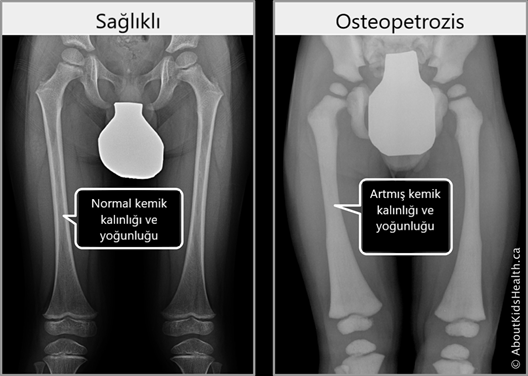
Tanı, klinik bulgulara ve çoğu zaman iskeletin radyografik görüntülenmesine dayanır. Hastalığın kesin tanısı için ise genetik testler uygulanabilmektedir.
Kaynak: https://www.aboutkidshealth.ca/Article?contentid=873&language=English
Tedavi Yöntemleri
Günümüzde osteopetrozise karşı etkili bir tedavi yöntemi yoktur. Uygulanan destekleyici tedaviler belirtileri azaltmaya yöneliktir.
Hastalığın Diğer İsimleri
Otozomal Dominant Osteopetrozis Tip 2, OPTA2
Kaynakça