Genel Tanı
Nonsendromik holoprozensefali, kafa ve yüz yapısını da etkileyen beyin gelişimi anomalisidir. Normalde beyin erken dönemdeki gelişimi sırasında ikiye bölünür (hemisferler). Holoprozensefali, beynin sağ ve sol hemisferlere ayrılacak şekilde bölünmediğinde ortaya çıkar. Bu duruma, genetik sendromların, kromozom anormalliklerinin veya doğumsal kusurlara neden olan maddelerin (teratojenler) neden olduğu diğer holoprozensefali tiplerinden ayırmak için nonsendromik denir. Nonsendromik holoprozensefali’nin şiddeti, aynı aile içindeki bireyler arasında bile değişkendir.Nonsendromik holoprozensefali, beynin bölünme derecesine göre dört tipte gruplanabilir. En şiddetli tipten hafif olana kadar sıralanacak olursa, tipler alobar, yarı-lobar, lobar ve orta interhemisferik varyant (MIHV) olarak bilinir.
Belirti ve Bulgular
Nonsendromik holoprozensefali’nin en şiddetli formlarında, beyin hiç bölünmez. Etkilenen bireylerde, bir merkezi göz (siklopi) (Şekil 1) ve gözün üzerinde yer alan tüp şeklinde bir burun yapısı (proboscis) (Şekil 1) vardır. Şiddetli Nonsendromik holoprozensefalisi olan bebeklerin çoğu, doğumdan önce veya hemen sonra ölür. Daha az şiddetli formlarda, beyin kısmen bölünür ve gözler genellikle birbirine yakındır (hipotelorizm) (Şekil 2). Hasta bireylerin yaşam süresi beklentileri, semptomların şiddetine bağlı olarak değişir.

Görüntü Kaynağı: https://www.humpath.com/IMG/jpg_holoprosencephaly_0301.jpg

Görüntü Kaynağı: https://ghr.nlm.nih.gov/art/large/hypotelorism.jpeg
Nonsendromik holoprozensefalisi olan insanlar genellikle küçük bir kafaya (mikrosefali) sahiptirler (Şekil 3), ancak beyinde artmış kafa büyüklüğüne (makrosefali) neden olan bir sıvı birikimi(hidrosefali) geliştirebilirler. Diğer bulgular arasında ağız tavanında bir açıklık (yarık damak) (Şekil 4) ve bu yarık damağa üst dudağın bir yarık ile ayrılması (yarık dudak) eşlik edebilir. İki yerine bir merkezi ön diş (tek bir merkezi maksiller incisor ) (Şekil 5) ve düz bir burun köprüsü de belirtiler arasında yer alabilir (Şekil 6). Göz küreleri anormal derecede küçük (microphthalmia) veya hiç oluşmamış (anoftalmi) olabilir.

Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/microcephaly.jpeg
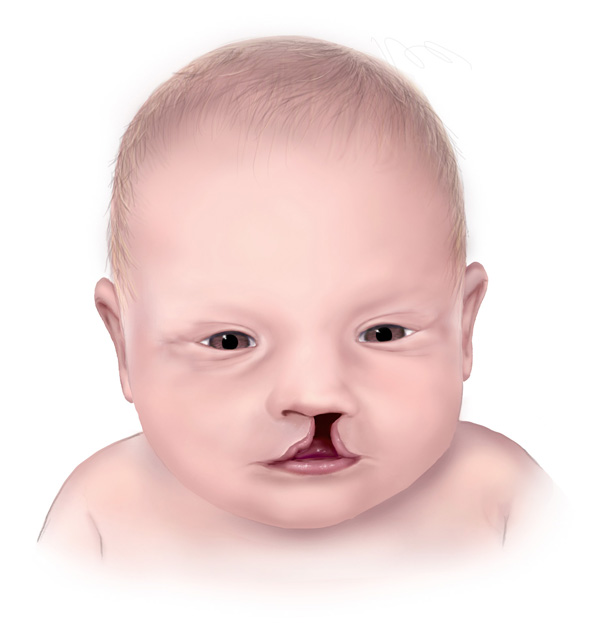
Görüntü kaynağı: https://www.cdc.gov/ncbddd/birthdefects/cleftlip.html

Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/single-maxillary-central-incisor.jpeg
Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/depressed-nasal-bridge.jpeg
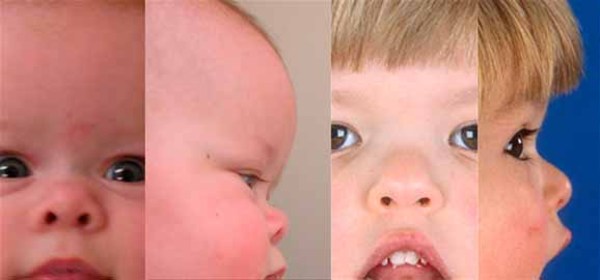
Nonsendromik holoprozensefalili bazı kişilerde başın şakak kısmında bir daralma , yukarı doğru bakan palpebral fissür , büyük kulaklar,kısa burun, ters dönmüş burun delikleri , burun ve ağız arasındaki boşluğun (philtrum) içe çökük ve geniş olması gibi fasiyal semptomlar olabilir. Genel olarak yüzdeki belirtilerin şiddeti, beyin anormalliklerinin şiddetiyle doğrudan ilişkilidir.Ancak, şiddetli fasiyal semptomları olmayan bireylerde de ciddi beyin anormallikleri olabilir. Bazı insanlarda görünür yapısal beyin anormallikleri yoktur ancak bu durumla ilişkili bazı yüz belirtilerine sahiptirler. Bu bireylerin mikroform holoprozensefali olarak bilinen bir bozukluğa sahip oldukları düşünülür ve tipik olarak ciddi şekilde etkilenmiş bir aile üyesinin doğumundan sonra tanı alırlar.
Nonsendromik holoprozensefalisi olan çoğu insanın gelişimsel olarak geriliği ve zihinsel kapasitesinde eksiklikler vardır. Etkilenen bireyler sıklıkla , çeşitli hormonlar üreten ve beynin tabanında yer alan, tam olarak işlevini yerine getiremeyen bir hipofiz bezine sahiptir. Hipofiz disfonksiyonu, hormonların kısmi veya tamamen yokluğuna yol açtığı için, çeşitli bozukluklara neden olabilir. Nonsendromik holoprozensefalisi ve hipofiz disfonksiyonu olan kişilerde en yaygın olarak, sıvı alımı ve idrar atılımı arasındaki dengeyi bozan diyabet insipidus görülür. Beynin diğer bölümlerindeki disfonksiyon; nöbetlere, beslenme güçlüklerine ve vücut ısısında, kalp atış hızında ve solunum düzeninde sorunlara neden olabilir. Eğer kokuyu algılayan beyin kısmı gelişmemiş veya eksikse, koku duyusu azalabilir (hiposmiye) veya tamamen yok olabilir (anosmia).
Görülme Sıklığı
Nonsendromik holoprozensefali, holoprozensefalili tüm vakaların yaklaşık olarak yüzde 25 ila 50’sini oluşturur ve bu da yaklaşık olarak 10,000 yenidoğanda 1’e denk gelmektedir.
Genetik Değişiklikler
Nonsendromik holoprozensefaliye neden olabilecek 14 tane gen bulunmuştur. Bu genler, özellikle beynin ve yüzün şeklini belirleyen ve normal embriyonik gelişim için önemli olan proteinlerin yapılması için gereklidir. Nonsendromik holoprozensefalisi olanların yaklaşık yüzde 25′ inde bu dört genden birinde bir mutasyon vardır: SHH, ZIC2, SIX3 veya TGIF1. Nonsendromik holoprozensefali ile ilişkili diğer genlerdeki mutasyonlar, olguların sadece küçük bir kısmında bulunur. Bu hastalığa sahip birçok bireyde tanımlanmış bir gen mutasyonu yoktur. Bu bireylerdeki bozukluğun nedeni bilinmemektedir.
Beyin, normalde hamileliğin üçüncü ila dördüncü haftasında sağ ve sol hemisferlere ayrılır. İki hemisfere ayıran çizgiyi kurmak için, birçok genin aktivitesi sıkı bir şekilde düzenlenmeli ve koordine edilmelidir. Bu genler; , sağ ve sol hemisferi oluşturacak sinyal proteinlerini yapmak için gerekli talimatları sağlar.
Sinyal proteinleri, gözlerin oluşumu için de çok önemlidir. Erken dönemdeki gelişim sırasında göze dönüşen hücreler, göz alanı olarak adlandırılan tek bir yapı oluştururlar. Bu yapı gelişmekte olan yüzün merkezinde yer almaktadır. SHH geninden üretilen sinyal proteini, göz alanının iki ayrı göze ayrılmasına neden olur. SIX3 geni, göz merceğinin ve retinanın oluşumunda görev alır.
Nonsendromik holoprozensefaliye neden olan genlerdeki mutasyonlar, anormal veya fonksiyonel olmayan sinyal proteinlerinin üretimine yol açar. Doğru sinyaller olmadan, gözler normal olarak oluşamaz ve beyin iki hemisfere ayrılamaz. Gözlerin doğru pozisyonuna geçmemesi durumunda yüzün diğer bölümlerinin gelişimi de etkilenir. Nonsendromik holoprozensefalinin belirti ve bulguları, beynin ve yüzün anormal gelişmesinden kaynaklanır.
Araştırmacılar, tespit edilmemiş olan diğer genetik veya çevresel faktörlerin, nonsendromik holoprozensefalinin şiddetini belirlemede rol oynadığına inanmaktadır.
Kalıtım Paterni
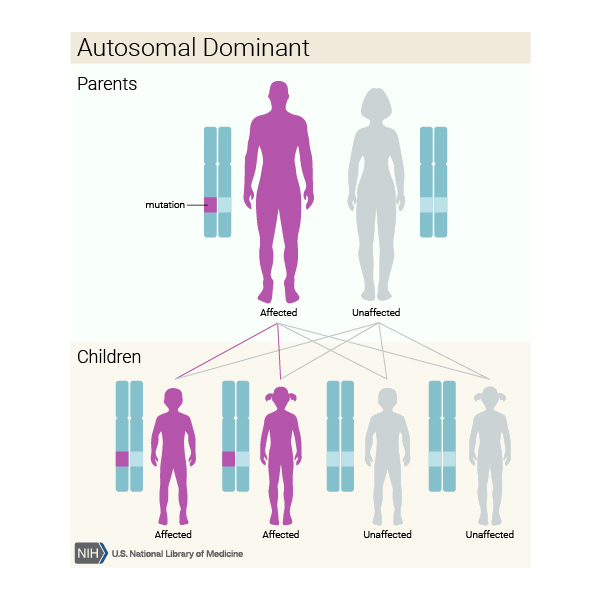
Nonsendromik holoprozensefali otozomal dominant kalıtılır, bu da her bir hücredeki bir genin, bozukluğa neden olmak için genellikle yeterli olduğu anlamına gelir. Bununla birlikte, bir gen mutasyonu olan tüm insanlarda belirti ve semptomlara rastlanılmaz.
Bazı vakalarda, etkilenen kişi, hastalığın özelliklerine sahip olan veya olmayan bir ebeveynin mutasyonunu miras alır. Diğer vakalar ise ailelerinde bozukluk öyküsü olmayan kişilerde, yeni bir gen mutasyonundan kaynaklanır.
Bilinen bir nedeni olmadan HPE’ si olan kişilerin aile üyelerinde hastalığa yakalanma riski genel olarak düşüktür.Sadece bazı durumlarda risk %50 civarı olmaktadır.
Tanı Yöntemleri
Çok ciddi vakalar, hamilelik sırasında sistematik ultrason taraması ve manyetik rezonans görüntüleme (MRG) ile tespit edilebilir. Daha sonraki dönemde tanı klinik özelliklere dayanmaktadır.
Ayırıcı Tanı
Ayırıcı tanıda anensefali, ağır konjenital hidrosefali, Walker-Warburg sendromu, geniş interhemisferik kist, otosefali ve diğer orta hat defektleri bulunur.
Antenal Dönemde Tanı
Yüksek klinik değişkenlik nedeniyle prenatal tanı, moleküler tanı yerine ultrason taramaları ve MRG’ ye dayanır.Moleküler tanı diyabetli annelerde ve aile öyküsü olanlarda yararlı olabilir.
Hastalıkla İlişkili Genler
Non sendromik HPE’ de, en az 14 gen etkilenmiştir: 4 majör gen (SHH (7q36), ZIC2 (13q32), SIX3 (2p21), TGIF (18p11)) ve 10 minör gen (PTCH1 (9q22) GLI2 (2q14), FOXH1 (8q24), TDGF1 (3p21), DISP1 (1q42), NODAL (10q22), FGF8 (10q24), GAS1 (9q21), DLL1 (6q27) ve CDON (11q23-q24)).tespit edilmiştir.
Hastalığın Diğer İsimleri
- holoprosencephaly sequence
- isolated holoprosencephaly
- isolated HPE
- non-syndromic, non-chromosomal holoprosencephaly
- non-syndromic, non-chromosomal HPE
- nonsyndromic HPE
HPE için Yurtdışındaki Kuruluşlar
- FACES: The National Craniofacial Association
- PO Box 11082
- Chattanooga, TN 37401
- Toll-free: 800-332-2373
- Telephone: 423-266-1632
- E-mail: [email protected]
- Website: http://www.faces-cranio.org/
- myFace
- 333 East 30th Street, Lobby Unit
- New York, NY 10016
- Telephone: (212) 263-6656
- E-mail: [email protected]
- Website: http://www.myface.org
Yararlanılan Kaynaklar
- https://ghr.nlm.nih.gov/condition/nonsyndromic-holoprosencephaly#
- https://www.omim.org/entry/236100?search=Holoprosencephaly&highlight=holoprosencephaly
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=301&Disease_Disease_Search_diseaseGroup=Holoprosencephaly&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Holoprosencephaly&title=Holoprosencephaly&search=Disease_Search_Simple
- https://rarediseases.info.nih.gov/diseases/6665/holoprosencephaly