Q
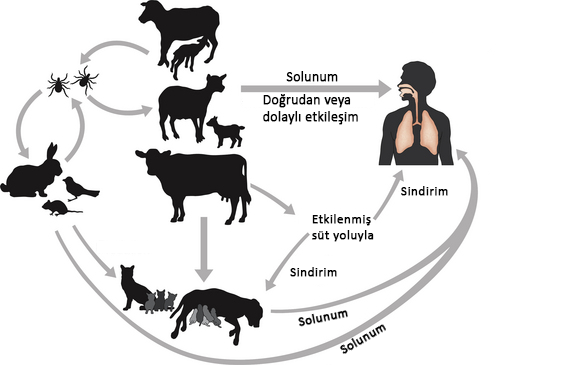
ateşi, Coxiella Burnetii bakterisinden kaynaklanan bir
hastalıktır. Genellikle keçi, koyun gibi hayvanlarda doğal olarak gözlemlenen
bu bakteri, solunum veya bakteriden etkilenmiş yemekleri yiyip içerek insanlara
geçebilir.
Etken Faktörler
Coxiella Burnetii bakterisinin bulaştığı hayvanlar; vucüt sıvılarıyla (doğum sırasında, dışkısıyla, sütüyle) veya hayvanın etinin yenmesiyle bakteriyi bulaştırabilir. Keneler bu hastalığın yayılmasında önemli faktörlerdir. Etkilenen hayvanlarla etkileşime geçen insanların hastalığa yakalanma ihtimali yüksektir.
Q
ateşi hastalığı genetik değildir. Bakteriyel bir hastalıktır.
Belirti ve Semptomlar
Q
ateşi hastalığının belirtileri kişiden kişiye değişiklik gösterebilir.
Hastalık, semptomsuz olabilir, aküt veya kronik olabilir.
Aküt
Q hastalığında, bakteriye maruz kalındıktan sonra 2-3 hafta içerisinde semptomlar
ortaya çıkar. Semptomlar arasında; ateş, yorgunluk, kas
ağrıları, mide bulanması, kusma, göğüs ve karın ağrısı, üşüme, terleme, ishal,
kilo kaybı ve kuru öksürük olabilir.
Hamilelerde düşük,
ölü doğum, erken doğum ve bebeğin gereğinden az kiloda doğması olabilir.
Ağır vakalarda ise
akciğer veya karaciğerde iltihaplanma görülebilir.
Kronik Q
hastalığı, bakterinin vucüda girmesinden aylar veya yıllar sonra belirti gösterebilir.
Genellikle, kalp ve damar hastalığı olan veya bağışıklık sistemi baskılanmış
hastalarda görülür. Görülme sıklığı %5ten azdır. Semptomları en çok
etkiledikleri bölgeye göre değişir, belirli bir semptomu yoktur. Uzun süreli
yorgunluk, ateş hali, eklem ağrılarına sebep olabilir.
Teşhis Yöntemleri ve Tedaviler
Q
hastalığının belirtileri genel olduğu için, kan tahliliyle tetkik edilmesi
gerekir. Özellikle hayvanlarla temasta olan hastalara, doktor kan tahlilinin
sonucunu beklemeden tedaviye başlayabilir. Antibiyotik genellikle ilk tercih
edilen tedavi şeklidir.
Aküt
Q hastalığı olan hastalarda, ilk üç günde antibiyotiğe başlamak en verimli
olur. Antibiyotiğin cevap vermediği durumlarda anti inflamatuvar ilaçlar
kullanılabilir.
Kronik
Q hastalığının tedavisi daha zordur, en çok etkilenen bölgeye göre kişiden
kişiye değişiklik gösterir. Bazı durumlarda ameliyata başvurulması gerekli
olabilir.
Akondroplazi, kısa ekstremiteli (uzuvlu)
cüceliktir. Akondroplazi, uzun kemiklerin kıkırdak içindeki kemik dokuya
dönüşmesini (osifikasyon) engelleyen bir rahatsızlıktır. Bu rahatsızlığa FGFR3
geninde yerleşik mutasyon (değişim) sebep olur. Hastaların% 80’inde
kendiliğinden (spontan) mutasyon görülken geride kalan% 20’inde hastalık
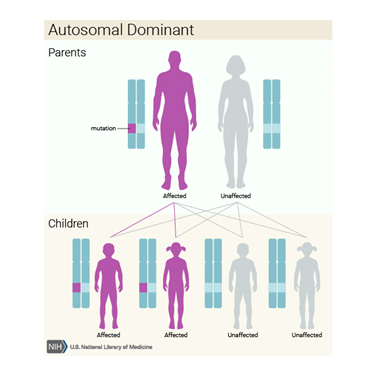
otozomal dominant olarak kalıtılır.
Belirti ve Özetler
Akondroplazi hastalığında belirtileri; kısa oğlan,
alışılmamış şekli büyük kafa (makrosefali), çıkıntılı alın, basık burun, kısa
kol ve uzakta, birinden uzaklanırken karın ve doğlar ve kısa ellerdir.
Akondroplazi hastası yeni doğanlarda kubbeli olduğu,
geniş alındığı görülür. Hidrosefali görülebilir. Bebeklik döneminde hipotoni,
akondroplazinin tipik bir şekildedir.
Genetik Görülme Sıklığı
Akondroplazi kadın ve erkeklerde eşit oranda görülür.
15.000-35.000 doğumda bir görülüyor.
Bazı popülasyonlarda, akondroplazinin görülme sıklığı
daha fazladır. Yıllar, Danimarka’da 6400 doğumda yaklaşık 1 vakada ve Latin
Amerika’da 10.000 doğumda yaklaşık 1 vakada meydana geldiği tahmin
edilmektedir. Ancak bir ırkın daha sık etkilendiği belgelenmemiş.
Kalıtım Paterni/ Deseni
Akondroplazi otozomal dominant olarak kalıtılır. Yani
hastalık temizleme için, genin bir ödenmelidir.
Teşhis Yöntemleri ve Tedaviler
Teşhis Yöntemleri
Akondroplazinin klinik ve radyolojik özellikleri iyi
durumda. Doğumdan hemen sonra klinik ve radyolojik olarak tanınabilir. Tipik
bulgulara sahip olanlardan birini tanıyı doğrulamak için moleküler genetik
testlere ihtiyaç duymazlar. Yeni doğanlarda tanıya dair bir şüphe uyandığında,
tanıyı doğrulamak için X-ışını (radyografi). FGFR3 geninde mutasyon var olup
olmadığına moleküler genetik testlerle bakılır.
Akondroplazi tanısında kullandığı klinik bulgular;
• Orantısız Kısa Erkek
• Makrosefali
• Orta yüz hipoplazisi ile tasarı dismorfik yüz
görünümü
• Dirsek ekstensiyonunda rekabet
• Kısa el ve ayak parmakları
• Lombar lordoz
• Çarpık yönetimi
Tedaviler
Bebeklerde foramen magnumun dekompresyonu ve
hidrosefali için şant gerekebilir.
Kulak tedavisi ve seröz otitis media tedavisi, işitme
problemlerinin yapılması ile birlikte. Konuşma terapisi önerilebilir. Çarpık
temizleme cerrahi ile düzeltilebilir. Lomber laminektomi gerekebilir.
Çocuklukta kilo alımı, daha sonra oluşabilecek komplikasyonları önlemek için
kontrol edilebilir. Boyundaki cini incitebilir aktivitelerden kaçınılabilir.
Sosyal ve psikolojik destek önerilebilir.
• Hidrosefalı: semptomların (hızlı kafa büyümesi,
görüşmede, baş ağrısı, şişkin alınabilir) ortaya çıkması, semptomlar (kafa
kafalarının büyümesi, kafa kafalarının büyümesi).
• Boyun ekleminde (kraniyoservikal bölge) daralma:
Ense dekompresyonu gerekebilir.
• Obstrüktif uyku apnesi: Kilo verme, bademcikleri ve
adenoidleri kapatma ameliyatı (adenotonsillektomi), pozitif hava yolu ve
nadiren boyun açıklığı için işleme (ameliyat) işlemi yapılabilir.
• Orta kulakta fonksiyon bozukluğu: Sıkı olabilir orta
kulak iltihabını ve olası işitme kaybını engellemek için, 7-8 yaşına kadar
kulak tüpleri vardır.
• Kısa oğlan: Büyüme hormonu kullanımı üzerine yapılan
araştırmalar, büyümenin başlangıçtaki hızılanmasını, ancak zamanla etkisi
azalır ve kalıcı faydalanabiliyor.
• Çarpık bacak: Ortopediste görünmek gerekir.
• Omurga Uzaklığı: Yaşamın ilk 12-18 liman
desteklenmemiş oturma yasağı dahil olmak üzere önleyici tedbirler, omurga
bölgesi (kifoz) sabit bir yerde eğri odada riskini içerir. Ciddiyet derecesine
bağlı olarak, bantlama veya cerrahi gerekebilir.
Hastalıkla İlişkili Genler
Bu hastalığa FGFR3 (fibroblast büyüme faktörü 3) adlı
gende oluşan mutasyon sebepleri olur. Bu gen, beyin ve kemik dokusunun
gelişimiinde görev alan bir proteini kodlar. Araştırmacılar, bu mutasyonların
FGFR3 proteinlerinin aşırı aktif olması nedeniyle neden olabilir, bunun için
iskelet gelişimini engellediğini ve bu rahatsızlıklara bakarken kemik
oluşumesinde rahatsızlıklara yol açtığını gösteriyor.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Galaktozemi, bireyin basit bir karbonhidrat
olan galaktozu bir başka karbonhidrat olan glikoza çevirmesini etkiler.
Özellikle süt ve süt ürünlerinde bulunan laktozun içerisinde, glikoz ve
galaktoz bulunur. Laktozlu ürün tüketilerek alınan galaktozun vucütta
kullanılabilmesi için glikoza çevirilmesi gerekir. Klasik galaktozemide bu çevirimi yapan GALT
(galactose-1-phosphate uridylyl transferase) enzimi eksikliği hastalığa sebep
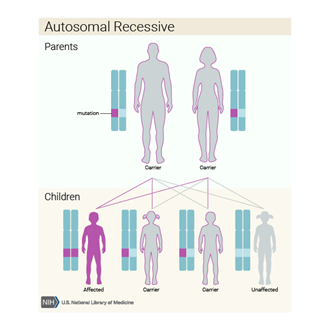
olur. Otozomal çekinik olan, kalıtımsal bir hastalıktır.
Belirti ve Semptomlar
Galaktozemi, birden fazla alt gruba
ayrılmıştır.
Klasik galaktozemi, hastalığın en yaygın ve
ağır şeklidir. Tip 1 olarak da bilinir. Yeni doğanlarda düşük galaktozlu diyet
hemen kullanılmazsa birkaç gün içerisinde komplikasyonlar meydana gelebilir.
Genellikle beslenme güçlüğü, kilo almada sorunlar, sarılık, karaciğerde büyüme,
kusma ve karında şişkinlik gözükür. Sonrasında ise ciddi bakteriyel
enfeksiyonlarla karşılaşılabilir. Hasta bireylerde katarakt, geç büyüme ve kız
çocuklarında yumurtalıkların erken fonksiyon kaybına yol açabilir.
Galaktozemi tip 2 ve tip 3 klasik tipten farklı
semptomlara yol açar ve görülme sıklıkları değişiklik gösterir.
Galaktozemi tip 2, galaktokinaz enzimi
eksikliğinden dolayı ortaya çıkar. Klasik tipe göre daha az saylık sorunu
yaşanır. Katarakt gözükebilir fakat uzun sğreli etkileri beklenmez.
Galaktozemi tip 3, galaktoz epimeraz eksikliği
olarak da geçer. Semptomların şiddeti değişiklik gösterebilir. Katarakt,
büyümede gecikme, zihinsel engel, karaciğer ve böbrek sorunlarına sebep
olabilir.
Genel olarak; galaktozun glikoza
çevirilmemesinden kaynaklanan vucüttaki fazla galaktoz, birikime sebep
olabilir. Bu birikim de karaciğerde büyümeye, siroz başlangıcına, karında asit
birikmesine, böbreklerde ve beyinde ciddi hasarlara sebep olabilir.
Genetik Görülme Sıklığı
Tip 1 Galaktozemi, 30.000-60.000 de 1
yenidoğanda görülebilir.
Tip 2 100.000de 1 den daha az görülürken tip 3
galaktozemi oldukça nadirdir.
Kalıtım Paterni/Deseni
Galaktozemi, otozomal çekinik bir hastalıktır.
Yani, iki genetik ebeveynden de hastalık geninin gelmesi gerekir. Aile
geçmişinde bu hastalık varsa gerekli genetik testler yapılmalı ve mutlaka önlem
alınmalıdır. Ayrıca, akraba evliliklerinde hastalığın görülme olasılığı
artacağından akraba evliliklerinden kaçınılmalıdır.
Teşhis Yöntemleri ve Tedaviler
Yenidoğanlarda topuktan alınan kan testinde galaktozemi
neredeyse %100 oranında belirlenebilmektedir. Yetkili doktor tarafından
galaktozemi teşhisi konulması gerekir.
Laktoz toleransı testi bu hastalara yapılmamalıdır.
Hastalığı tamamen ortadan kaldıracak bir tedavi yöntemi
şimdilik bilinmemektedir. Bu hastalığa uygun beslenme düzeniyle semptomlar en
az seviyeye çekilebilir, hatta kaldırılabilir. Beslenme düzeninden laktoz ve
galaktoz bulunan tüm ürünler çıkartılmalıdır. Hayatı boyunca bu bireyin
beslenme düzeninin laktozsuz olacağının kabullenilmesi gerekir.
Hastalıkla İlişkili Genler
Klasik galaktozemi, GALT geni sayesinde
üretilen GALT enziminin yokluğu veya düzgün çalışmamasından ortaya çıkan bir
hastalıktır.
İyonize radyasyona maruz kalma sonucu akut, gecikmiş olarak
veya kronik olarak etkilerini gösteren bir hastalıktır. Büyük dozlarda etkiler
hemen ortaya çıkarken, küçük dozlarda radyasyonun vücutta birikmesi sonucu
genetik ve uzun süreli etkiler görülmektedir. Radyasyondan zarar görüp hasara
uğrayan hücrelerin tamiri için tedavi bulunmamaktadır. Ancak yakın zamanda FDA (Amerikan
Gıda ve İlaç Dairesi), radyoaktif elementlerin vücuttan atılmasında etkili olan
ilaçların onayını vermiştir. Hücre hasarı geri dönüşsüzdür ancak bu ilaçlarla
hastalarda radyasyona maruz kalma sonucu çıkan belirtiler tedavi
edilebilmektedir.
İlk Radyasyon Hastalığı vakaları Hiroshima ve Nagasaki
nükleer patlamalarından sonra görülmüş, Japon hekimler bu hastalığı “gözle
görülür hiçbir hasar olmadan, birden ortaya çıkan hastalık” olarak
tanımlamıştır. Günümüzde, bu hastaların radyasyon hastalığına yakalandığı anlaşılmıştır.
Düşük miktarda maruziyet sonucu bu hastalık soğuk algınlığında görülen hafif
belirtilerle hastayı terk edebiliyorken; yüksek miktarda maruziyet Chernobyl
patlamasında olduğu gibi hastada ölümcül etkiler yaratabilir.
Toplam doz ve doz oranı radyasyonun somatik ve genetik
etkilerini belirler. Radyasyon dozu belirtilirken üç ölçü birimi kullanılır:
Roentgen, rad ve rem. Roentgen (R), havadaki x ve gamma ışınlarının miktarını
belirtirken; rad (radiation absorbed dose), tüm radyasyon tiplerinde absorbe
edilen enerji miktarı için kullanılır. Rem ise nötronlar gibi bazı radyasyon
türlerinin, eşdeğer miktarda emilen enerji için ne kadar biyolojik etki
üretebileceğini gözlemlemek için kullanılır.
Hastalığın Diğer İsimleri
null
Radiation Disease
Radiation Effects
Radiation Illness
Radiation Injuries
Radiation Reaction
Radiation Syndrome
Belirti ve Semptomlar
Akut radyasyon hastalığı bulantı, kusma, ishal, anoreksiya,
baş ağrısı, halsizlik ve taşikardi (kalp çarpıntısı) belirtileriyle kendini
gösterir. Hafif olgularda belirtiler birkaç saat veya gün içinde kaybolur. Akut
radyasyon hastalığının yüksek doz veya düşük doza bağlı olarak ortaya çıkan çeşitli
tipleri vardır.
Akut radyasyon hastalığı çeşitleri doz, doz oranı, maruz
kalan vücut bölümü ve maruziyetten sonra vücuttan atılması için geçen süreye
göre hastada gözlemlenmektedir. Radyasyona
maruziyet sonucu dakikalar içinde radyasyon tüm vücudu etkiler.
Akut Radyasyon hastalığı üç aşamayla gelişir: İlk aşama
birkaç dakikadan birkaç güne kadar sürebilir ve hastada mide bulantısı, diyare
ve kusma görülür. Bundan sonraki aşamada hasta birkaç hafta iyileşme
belirtileri gösterir. Son aşamada ise hastadan hastaya değişmekle beraber
kardiyovasküler, gastrointestinal, hematopoietik veya sinir sistemi
rahatsızlıkları görülmektedir. Yüksek dozda (3000 rad’dan fazla radyasyon
maruziyet) birkaç saat içinde ölümcül kardiyovasküler belirtiler ve sinir
sistemi rahatsızlıkları, mide bulantısı, kusma, anksiyete (endişe), konfüzyon
(bilinç bulanıklığı) ve bilinç kaybı görülmektedir. 5-6 saat sonunda tremor
(titreme) ve konvülziyon (havale) başlar ve üç gün sonunda ölüme sebep olur. 400
rad’dan fazla radyasyona maruz kalma sonucu mide bulantısı, kusma, elektrolit
dengesizliği, diyare (ishal), sıvı kaybı, plazma hacminde azalma gibi
gastrointestinal rahatsızlıklar ortaya çıkar. 200-1000 rad arasında, 6 ila 12
saatlik radyasyon maruziyeti sonucunda ise anoreksiya, ateş, halsizlikle
seyreden hematopoietik rahatsızlıklar oluşur.
Nedenleri
İyonize radyasyonun en büyük kaynağı özellikle kanser tedavisinde kullanılan yüksek enerjili x-ray ışınları ve radyum vb. radyoaktif elementlerdir. Bunun dışında nükleer santrallerde meydana gelen patlamalar veya nükleer silah savaşları da başlıca hastalık kaynaklarıdır. Örneğin, Hiroshima, Nagasaki ve Chernobyl nükleer santrallerinde patlamalar sonucunda bu bölgeler ve çevre bölgelerdeki insanlarda kanser, mutasyon ve genetik hasarlar meydana geldi; uzun yıllar boyunca radyasyon maruziyeti ve etkileri devam etti.
Vücudun radyasyona maruz kalan kısmı bu hastalığın ortaya
çıkmasında önemli bir faktördür. İnsan vücudu 200 rad’a kadar radyasyonu
ölümcül risk olmadan absorbe edebilmektedir. Radyasyon dozu 450 rad’a
ulaştığında ölüm riski %50’ye ulaşırken bazen 600 rad kısa sürede ölüme sebep
olabilir.
Radyasyonun vücutta dağılımı da hastalığın seyrini etkileyen
başka bir faktördür: Örneğin bağırsak ve kemik iliğinin korunması hastanın
ölümünü engeller.
Etkilediği popülasyon
Radyasyon hastalığı kadın ve erkekleri eşit oranda
etkilemektedir.
Teşhis
Teşhis, radyasyon maruziyeti öyküsüne bağlı olarak
belirlenir. Maruz kalma ve kusma arasında geçen süre maruziyet seviyesinin
tespitinde önem teşkil eder.
Maruz kalan hastaların Geiger sayacı veya tüm vücut analizi
için kullanılan sayaçlarla izlenmesi gerekmektedir.
Tedavi
Radyoaktif elementlerin deriden maruziyeti sonucu derhal
kontaminasyon (uzaklaştırma) sağlanmalıdır. Hasta bol sıvıyla durulanmalı ve
EDTA (radyoaktif izotopların etkisini indirgemek için kullanılan bir madde) vb.
maddeler uygulanmalıdır. Ayrıca küçük yaralar ve maruz kalan dokular da
kontaminasyonu engellemek için iyice temizlenmelidir. Eğer radyasyon içeren bir
madde ağız yoluyla vücuda alınırsa kusturma veya mide yıkaması
yapılmalıdır.
2015 yılında radyasyonun kemik iliğine sebep olduğu yetişkin
ve çocuklarda kullanılmak üzere Amgen firması tarafından Neupogen adlı bir ilaç
piyasaya sürüldü. Bunun haricinde radyoaktif talyum maruziyetinin tedavisinde,
FDA onaylı aslında endüstride kullanılan Prusya mavisi adında bir pigment
kullanılmaktadır. Vücutta bu elementlerin absorblanmasını engelleyerek etki
göstermektedir. Plütonyum, amerikyum ve bakır radyoaktiflerine karşı Ca-DTPA ve
Zn-DTPA kullanılması FDA tarafından onaylanmıştır.
Genel Bilgi, Genetik
Değişiklikler/Etken Faktörler
TANGO2, 22. Kromozomda (22q11.21) bulunan
protein kodlayan bir gendir. Bu gendeki mutasyon, otozomal çekinik olarak
gelecek nesle aktarılır. Etkilenen bireyler, metabolik kriz de denilebilen
aralıklı akut rahatsızlıklara yakalanabilir. Uzun süre açlık veya nexle gibi
bir başka bu krizleri tetikleyebilir. Metabolik krizler sırasında aritmiler
(kalp atışı ritminin anormal değişimi), rabdomiyoliz (iskelet kası dokusundaki
hasar sebebiyle bozulma) veya
ensefalopati (beyinde oluşan hasar, arıza) meydana gelebilir. Her birey bu
hastalığı farklı deneyimleyebilir.
Bilinen bir tedavisi yoktur
fakat araştırmalar devam etmektedir.Hastalığın etkilerini en aza indirgemek ve kontrol
altında tutmak şimdilik asıl amaçtır.
Görülme sıklığı
Bu hastalık literature 2016 yılında girmiştir. TANGO2
Research Foundation tarafından hazırlanan rapora göre 2018 Mayısı itibariyle
bilinen vaka sayısı dünyada 30 bireyden azdır.
Belirti ve Semptomlar
Hastalığın az görülmesi ve bireyler arasında farklılık
göstermesi nedeniyle tanı kriterleri kesinleştirilmemiştir. Semptomlar arasında
gelişimde gecikmeler, sakarlık, hipotiroid, koordinasyonda bozukluklar, atak
geçirme, düşük kan şekeri, toksik maddelerin kanda artmasıyla ilgili
rahatsızlıklar ve hastalığın çok geliştiği evrelerde bilinç kaybı yaşanabilir.
Ayrıca, kas dokusunun bozulmasıyla kaslarda ağrı, zayıflık
ve yorgunluk gözükebilir. Doku bozulması, kreatinin kinaz ve miyoglobinin kana
karışmasını sağlayabilir. Bu durumda, miyoglobin böbreklerde birikmeye,
sonrasında da böbreklerin iflas etmesine neden olabilir.
Ayrıca, aritmilere neden olabileceğinden uzun süreli kalp
atışının değişikliği, kalp ve damar hastalıklarına sebebiyet verebilir.
Belirtileri daha da sıralamak mümkündür fakat TANGO2ye bağlı hastalığın olması,
bu belirtilerin kesin gösterileceği anlamına gelmez. Yetkili bir doktora
başvurmak gerekir.
Kalıtım Paterni/Deseni
Otozomal çekinik bir hastalıktır. Eğer bireyin
iki genetic ebeveyni de bu hastalığın taşıyıcısıysa, %25 oranında bu hastalık
kendini gösterebilir. %50 oranında birey, bu hastalığın taşıyıcısı olur ve %25
oranında da hastalıktan etkilenmemiştir/taşıyıcı değildir.
Bununla birlikte, TANGO2 genindeki herhangi bir
mutasyon da farklı bir hastalık veya fonksiyon bozukluğu sağlayabilir.
Araştırmalar devam etmektedir.
Teşhis Yöntemleri ve Tedaviler
Teşhis için yetkili bir doktora ve genetik
teste ihtiyaç vardır. TANGO2 genindeki genetic değişim, bu hastalığın teşhisi
için önemli bir kanıttır. Aile geçmişinde bu hastalık var ise, doğum öncesi
genetic testlere de başvurulabilir.
Hastalığın kontrol altında tutulması ve etkilerinin
azaltılması için metabolic krizler önceden fark edilmeli ve önlem alınmalıdır.
Önlemler kişiden kişiye değişir, genel olarak uzun süreli açlığın ve susuzluğun
önlenmesi gerekir. Tedavi yöntemleri bu hastalığı iyileştirmekten çok
semptomları azaltmayı ve kontrol altında tutmayı amaçlar. Örneğin bir bireyde
başlıca sorun TANGO2 ye bağlı bu hastalığın aritmiye sebep olması ise; aritmi
için tedavi yöntemleri uygulanır.
Hastalıkla İlişkili Genler
TANGO2, taşıma ve golgi aygıtına bağlı bir gendir. Kodladığı proteinin tam işlevi şu anlık bilinmemektedir. Araştırmalar devam etmektedir.
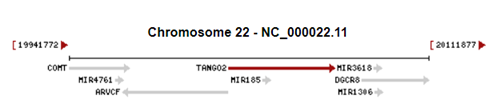
Fotoğraf1. TANGO2 geninin 22. Kromozomdaki yeri
fotoğrafta kırmızı okla belirtilmiştir.
Laband sendromu oldukça nadir
görülen genetik hastalıktır ve aynı zamanda Zimmerman-Laband sendromu olarakta
adlandırılır. Laband sendromu kafa, yüz, eller ve ayaklarda gözlemlenen
anormallikler karakterize edilir. Laband sendromuna sahip çoğu çocukta normal
olmayan büyüklükte diş etleri gözlemlenir ve bu sebeple çiğneme, yutma, veya
konuşma kabiliyetleri etkilenebilir. Ek olarak, Laband sendromlu yeni
doğanlarda oldukça uzun, ince el ve ayak parmakları, eksik ya da şekilsiz
tırnaklar gözlemlenebilir. Bazı rapor edilen vakalarda aynı zamanda zihinsel
gerilikte gözlemlenmiştir. Laband sendromu çoğu vakada otozomal dominant olarak
aktarıldığı bulunmuştur. Fakat, otozomal çekinik olarak aktarıldığı vakalarda
gözlemlenmiştir.
Belirti ve
Semptomlar
Genel bilgide de bahsedildiği üzere
laband sendromu oldukça nadir hastalıktır. Yüz, kafa, eller ve ayaklarda
anormallikler görülür. Laband sendromlu bebeklerde doğuştan itibaren gelen el
ve ayak parmaklarında anormal yapılar gözlemlenebilir. Diğer semptomlar,
ilerleyen çocukluk dönemlerine kadar gözlemlenmeyebilir.
Laband sendromlu çoğu çocukta
gereğinden büyük diş etlerinin varlığı gözlemlenebilir. Diş etlerinin aşırı
büyümesi farklı semptomlara da neden olur. Bunlardan birkaç tanesi ise dişlerin
çıkmasının gecikmesi, dişlerin birbiriyle olması gerektiği gibi yan yana
olmamasından dolayı kusurlu bir diş kapanışı olarak sıralanabilir. Sadece
bunlarla da kalmayıp çiğneme problemleriyle birlikte konuşma sıkıntıları, salya
artışına veya ağızın anormal bir şekilde fazla kurumasına, yutkunmada
zorluklara, ağız kenarlarında yara oluşumuna, dişlerin erken kaybına da sebep
olabilir. Bazı uç vakalarda ise diş etlerinin bütün dişleri kaplayacak kadar
büyümesi gözlemlenmiştir.
Laband sendromuna sahip bireylerde
yukarıda da bahsedilen yüz anormallikleri erken çocukluk döneminde ortaya çıkıp
ergen boyunca anormallikler ağırlaşabilir. Bahsi geçen anormalliklerin içinde
dar bir yüz yapısı, aşırı büyümüş dil, dudaklar, burun, kulaklardan herhangi
bir veya birden fazlası olabilir. Kulaktaki ve burundaki kıkırdak yapıları
Laband sendromlu bireyler normalden daha yumuşak olması gözlemlenebilir ve ek
olarak bazı Laband sendromlu çocuklarda aşırı saç uzaması da gözlemlenmiştir.
Çoğu Laband vakalarında el ve
ayaklarda şekil bozuklukları gözlemlenebilir. Şekil bozuklukları oldukça uzun
ve ince el, ayak parmakları (araknodaktili) ile beraber şişik parmak uçları
içerebilir. Bazı vakalarda ise el, ayak şekil bozukları parmak uçlarının
kusurlu oluşumunu, el ile el parmaklarının eklemlerinin olağandışı bir seviyede
esnek olmasını içermiştir. Ek olarak, el ve ayak tırnaklarında, özellikle baş
parmaklarda, kusurlu oluşum veya oluşmama gözlemlenebilir.
Bazı Laband sendromlu bireylerde
farklı fiziksel semptomlarda gözlemlenmiştir. Bazılarında yukarıda
bahsedilenlere ek olarak iskeletsel ve omurgasal anormallikler, oldukça büyük
karaciğer gözlemlenebilir. Normal zeka seviyesi bazı vakalarda gözlemlenirken
bazılarında ise zeka geriliği gözlemlenmiştir.
Genetik Görülme
Sıklığı
Laband sendromu aşırı nadir genetik bir hastalıktır. Ek
olarak, dişi ve erkekleri etkileme oranı olarak bir farklılık
gözlemlenmemiştir. 1928’de hastalık ilk tanımlandığından beri literatürde 30
farklı vaka bulunmuştur.
Kalıtım
Deseni
Çoğunlukla Laband sendromu otozomal dominant olarak
kalıtıldığı gözlemlenirken azınlıkta olarak otozomal çekinik olarak ta
aktarıldığı rastlanmıştır.
Teşhis
Yöntemleri ve Tedaviler
Laband sendromu ile sonuçlanan çoğu
teşhis erken çocukluk döneminde olduğu fark edilir. Teşhisten özenli bir klinik
inceleme ile ayrıntılı bir hasta geçmişi, özelleştirilmiş testler, el ve ayak
parmaklarının röntgen görüntüsü, burnun, kulakların, dudakların ve dilin
ayrıntılı incelenmesi beraber doğrulama yolu takip edilir. Kesin doğrulanma
yolu olan aşırı büyümüş diş etleri olması süt dişlerinin çıkışına kadar
gözlemlenmeyebilir.
Laband sendromlu bireyin tedavisi
kişide gözlemlenen semptomları tedavi etme yolundadır. Kapsamlı bir tedavi
pediatri uzmanlarını, diş anormalliklerini incelemek ve düzeltmek üzerinde
uzmanlar (dişçiler, ortodonti uzmanları), kemiksel anormallikleri düzelten
uzmanlar (ortopedi uzmanları), diş eti hastalıkları uzmanı (periodontist),
farklı mesleklerden uzmanların beraber çalışmasını gerektirebilir.
Ek olarak bahsetmek gerekir ki bazı
vakalarda doğru dürüst uygulanan ağız hijyeni dişetlerinde oluşan
anormallikleri oluşumunu geçiktirdiği veya azalttığı gözlemlenmiştir.
Dişetindeki anormallikler bazı vakalarda ameliyatsal olarak tedavi edilebildiği
de gözlemlenmiştir. Fakat, bunlara rağmen diş etlerinin aşırı büyümesi hala
görüldüğü vakalarda olmuştur. Diş etlerinin dişleri tamamen kapattığı
durumlarda düzgün bir ağız hijyeni sağlamak hasta için oldukça zorlaşabildiği
gözlemlenmiştir.
Semptomlarda özet geçildiği üzere
Laband sendromlu çocuklarda karaciğer veya dalak büyümesi riski olduğundan
dolayı erken teşhis çocuklar başta olmak üzere oldukça önemlidir.
Pitt-Hopkins
sendromu zihinsel engellilik, gelişimsel gecikme, solunum problemleri, nöbetler
(epilepsi), tipik yüz yapısı ve yüksek miyopi özellikleri ile karakterize olan
genetik bir sendromdur. TCF4 geninin mutasyonu sonucu ortaya çıkar ve otozomal
dominant karakterdedir. İlk defa 1978 yılında David Pitt ve Ian Hopkins
tarafından tanımlanmıştır. Nedeni bilinse de tedavisi için bir ilacı
bulunmamaktadır.
Genetik Değişiklikler/Etken Faktörler
Pitt-Hopkins
sendromuna TCF4 genindeki mutasyon ya da 18. kromozomun TCF4 genini içeren
bölgesindeki delesyon sebep olmaktadır. Otozomal baskın (dominant) olarak
kalıtılır fakat ailesinde Pitt-Hopkins sendromu öyküsü olmadığı halde de
mutasyon sonucu ortaya çıkmaktadır.
TCF4
geni Pitt-Hopkins sendromundan başka Şizofreni, Otizm, Fuchs Kornea Distrofisi
ve Karaciğer hastalığı olmak üzere çeşitli hastalıklarda da rol oynar.
Belirti ve Semptomlar
Pitt-Hopkins
sendromunun birçok belirti ve semptomu bulunmaktadır. Semptomlar ve şiddetleri
kişiden kişiye değişebilir. Erken semptomlar bebek doğduktan sonra bir sene
içinde görülmektedir. Çok düşük kas tonusu (hipotoni) ve gelişim geriliği
görülmektedir. Bazı bebeklerde baş boyutu küçüktür (mikrosefali). Çocuklar
yürümeye beklenenden aylar hatta yıllar sonra başlayabilir. Bazıları ise
bağımsız olarak yürüme yeteneğini kazanamazlar. Konuşma gecikmiştir. Bazıları
birkaç kelime söylemeyi öğrenebilirken çoğu konuşamaz. Bununla birlikte
bazıları basit yönergeleri anlayıp uygulayabilir. Zihinsel engellilik seviyesi
genellikle orta ila şiddetlidir.
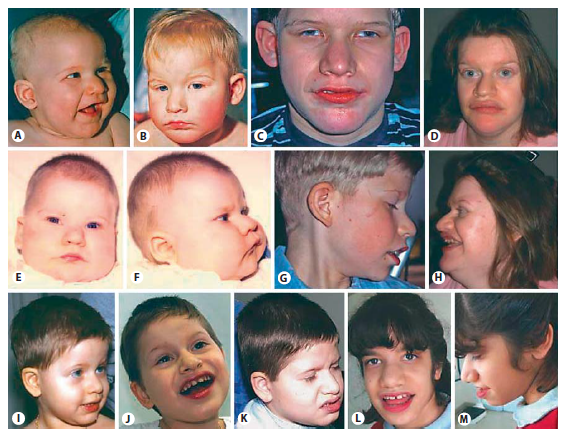
Yüz özellikleri çukur gözler, çıkık burun, aşağı yönelmiş burun ucu, geniş burun delikleri ve burun kemeri, kısa filtrum, geniş ağız, geniş aralıklı dişler ve çıkık çene ile ayırt edilir. Bu özellikler yaş ile daha belirgin hale gelir. (Şekil 1)
Şekil 1. Pitt-Hopkins sendromu hastalarında yüz özellikleri. Hasta 1 A (6 aylık), B (18 aylık) ve C (14 yaş). Hasta 6 D ve H (29 yaş). Hasta 2 E, F (6 aylık) ve G (11 yaş). Hasta 3 I ( 3 yaş), J (6 yaş) ve K (8 yaş). Hasta 4 L ve M (12.5 yaş).
Otizm
spektrum bozukluğu belirtileri görülebilir. Beslenme sırasında alışılmadık davranışlar,
agresif davranışlar, anksiyete, stereotipik el ve kafa hareketleri görülür. Çoğu
çocukta mutlu bir yüz ifadesi vardır. Uykuda kaybolan, soluk alıp verme
problemi görülür. Bunlar genelde anksiyete, heyecanlanma ya da yorulma sonucu
gerçekleşir. Hızlı nefes alıp verme ardından nefes almamaya veya nefes almaya
çalışma davranışları birbirini takip eder. Pitt-Hopkins sendromuna sahip
kişilerin neredeyse yarısında nöbet görülür. Nöbetler çocukluk ya da gençlik
döneminde herhangi bir zamanda başlayabilir. Uyuya kalma ya da uykusuzluk gibi
sorunlar görülebilir. Hastaların yarısından azında gastroözofageal reflu
görülür. Miyopi iki yaşından önce görülebilir ve şiddetli olabilir. Bunun
haricinde şaşılık ve astigmatizm de görülür. İskelet sisteminde skolyoz,
düztabanlık, yumru ayak, küçük el ve ayaklar, kavisli ya da bükük parmaklar,
geniş parmak uçları ya da gittikçe incelen parmaklar görülebildiği rapor
edilmiştir. Ayak parmakları üst üste binmiş halde olabilir. Acı eşikleri
yüksektir. Etkilenen erkeklerin yaklaşık üçte birinde testislerden biri ya da
her ikisi de skrotuma inmemiştir.
Genetik Görülme Sıklığı
Pitt-Hopkins sendromunun oldukça
nadir olduğu düşünülmektedir. Dünya çapında 500 kişinin bu sendroma sahip
olduğu rapor edilmiştir. Kadın ve erkekte görülme sıklığı eşittir.
Kalıtım Paterni/Deseni
Otozomal
baskın (dominant) olarak kalıtılır. Baskın genetik hastalıkların ortaya çıkması
için anormal genden bir tane bulunması yeterlidir. Bu gen anne veya babadan
gelmiş olabilir.
Üreme
hattı mozaikliği görülebilir. Bu durumda anne veya baba TCF4 gen mutasyonunu
vücut hücrelerinde taşımaz yani etkilenmemiştir. Fakat üreme hücreleri (yumurta
ya da sperm) mutasyona sahiptir. Yumurta veya spermlerden biri mutasyona
sahipse hastalık yavru bireyde görülür. Ebeveynlerin kanlarından yapılan testte
mutasyon negatif çıksa bile sonraki çocuklarının da etkilenmiş olma
olasılıkları yaklaşık %1-2 kadardır.
Teşhis Yöntemleri ve Tedaviler
Pitt-Hopkins
sendromunun teşhisi detaylı hasta hikayesi, klinik bulgular ve semptomun
karakteristik özelliklerinin tanımlanmasıyla gerçekleştirilir. Ancak bu sendrom
ve diğer bazı nörolojik bozukluklarda görülen semptomlar benzerlik
göstermektedir. EEG ve beyin MRI görüntülemesiyle ve genetik analizler
sonucunda TCF4 geninde meydana gelmiş mutasyon ya da delesyon tespit edilerek
teşhis desteklenir.
Bilinen
bir tedavisi yoktur. Tedaviler semptomları gidermek için uygulanır.
Hastalıkla İlişkili Genler
TCF4
genindeki mutasyonlar Pitt-Hopkins sendromuna neden olmaktadır. Bu gen 18.kromozomun
uzun kolunda bulunmaktadır (18q21.2). TCF4 geni, vücutta birçok fonksiyonda
kritik rol oynayan proteinlerin yapımı için bilgiler içerir. Bu gende mutasyon
gerçekleştiğinde oluşan protein hatalı ve etkisiz olabilir, eksik veya fazla
üretilebilir. TCF4 geni, transkripsiyon faktörü olarak da bilinen TCF4
proteinini sentezlenmesini sağlar. Bu proteinin farklı gelişim evrelerinde hücre
farklılaşması ve hücre ölümü gerçekleştirmek gibi önemli görevleri vardır.
İnsan gelişiminin erken dönemlerinde sinir sistemi gelişimiyle ilişkili olarak
yüksek düzeyde eksprese edilir.
Hastalığın Diğer İsimleri
Hastalık
PHS ve PTHS kısaltmalarıyla da adlandırılır.
Peippo M., Ignatius J. Pitt-Hopkins Syndrome. Mol Syndromol
2011;2:171–180
Sweatt
J. D. Pitt–Hopkins Syndrome: intellectual disability due to loss of
TCF4-regulated gene transcription. Experimental & Molecular Medicine (2013)
45
Oculodentodigital Displazi, özellikle gözleri (oculo),
dişleri (dento) ve parmakları (digital) etkileyen sistmik bir rahatsızlıktır.
Küçük gözler, görme kaybı, kayıp dişler, tekrarlayan diş çürükleri ve
parmaklardaki fazladan kemiksi büyümeler gibi semptomlar verebilir. GJA1 geninde mevcut bir mutasyon sonucu
görülebilecek bu displazinin otozomal dominant kalıtıldığı düşünülmektedir.
Oculodentodigital Displazi, fizik muayene ile teşhis edilip, genetik testlerle
doğrulanabilir. Hastalığın idare edilmesinde semptomlara yönelik tedaviler
kullanılabilir. Aynı zamanda erken teşhisle hayat kalitesi arttırılabilir.
Genetik
Değişikler/ Etken Faktörler
GJA1
genindeki (6.
Kromozomun uzun kolunda: 6q21-q23.2) mutasyonlar Oculodentodigital
Displaziye yol açmaktadır. GJA1 geni,
Connexin 43 proteini yapımından sorumludur. Bu protein, hücreler arası direkt
iletişimi sağlayan aralıklı bağlantılar için bir alt ünite üretir. Connexin 43
tarafından üretilmiş aralıklı bağlantılar vücudun çeşitli yerlerinde bulunur.
GJA1
genindeki
bir mutasyon, anormal Connexin 43 üretimine sebep olur. Bu anormal proteinle
oluşmuş kanallar genellikle kapalıdır. Bazı mutasyonlar sonucunda, Connexin 43
kanal oluşturması gereken hücre yüzeyine ulaşma yetisini kaybeder. Bu sebeple
hücreler arası iletişim bozulur ve sonucunda hücrelerin büyüme ve özelleşme
süreçlerinde hatalar olur. Oculodentodigital Displazi’de görülen morfolojik
bozuklukların sebebi de bu hatalardır.
Belirti
ve Semptomlar
Özellikle
4. ve 5. parmaklar arası yapışıklık (sindaktil)
İncelmiş,
hassas diş minesi, buna bağlı olarak eksik dişler ve tekrarlayan kaviteler
Ucu
ince, dar burun delikleri ve dış kanatları incelmiş spesifik bir burun
Yavaş
uzayan, kuru saçlar (hipotrikoz)
Araları
oldukça açık, küçük gözler (mikroftalmi)
Glokom
Şaşılık
(Strabismus)
Görme
kaybı
Mikrosefali
Yarık
damak
Çeşitli
nörolojik semptomlar ( Ataksi, kaslarda spastisite, duyma kaybı, idrar veya
gayta inkontinansı, disartri)
Palmoplantar
Keratoderma
Alt
çenede fazla gelişme
Kafatası
kemiklerinde anormal kalınlaşma
Anormal
genişlikte köprücük kemikleri
Kulak
memelerinde anormal kalsiyum birikmeleri
Kardiyak
malformasyonlar
Genetik
Görülme Sıklığı
Oculodentodigital Displazi kadınları ve erkekleri eşit
sıklıkta etkileyen oldukça nadir bir hastalıktır. Medikal literatüre geçmiş 85
vaka bulunmaktadır. Otozomal resesif formu yalnızca 5 vakada saptanmıştır.
İnsidans tam olarak bilinmemektedir. Çoğu vakanın teşhissiz kaldığı düşünülmektedir.
Kalıtım
Paterni
Oculodentodigital Displazi, çoğunlukla otozomal
dominant olarak kalıtılmaktadır. Ancak literatürde aile öyküsü olmadan yeni
mutasyonlarla oluşmuş bireyler de mevcuttur.
Teşhis
Yöntemleri/ Tedaviler
Teşhis, fizik muayene ve genetik testlerle mümkündür.
Tedavi ise semptomlara yöneliktir.
N-Acetylglutamate
Sentaz (NAGS) Eksiliği otozomal çekinik gen ile taşınan, hiperamonyemiye neden
olan üre siklüs bozunluklarındandır. Bu hastalık hem vücudun proteinleri
işlemesini hem de vücuttan amonyak atılımını etkileyen metabolik bir
bozukluktur. Hastalığın sonucu olarak kişi vücudundan amonyağı atamaz ve bunun
sonucu olarak vücutta fazlaca amonyak birikir. Amonyak, vücudun proteinleri
işlemesiyle ortaya çıkar ve vücutta çokça birikirse kişi için zehirli hale
gelir.
Epidemiyoloji
Çok
nadir ve tam olarak yaygınlığı bilinmemektedir. Ancak dünya genelinde 1-2
milyon insanın bu hastalığa sahip olduğu tahmin edilmektedir.
Klinik Bilgi
Hastalık her yaşta ortaya
çıkabilmektedir, ancak çoğunlukla yenidoğanlarda görülmektedir. Klinik görünümü
değişiklik göstermekle beraber en yaygın olanları; kusma, hiperaktivite,
letarji (uyuşukluk), diyare (ishal), beslenmede isteksizlik, nöbet, hipotoni,
geç psikomotor gelişimi ve solunum bozukluklarıdır. Hiperamonyemi sonuçları ağır olan bir hastalıktır ve çoğunlukla
hiperamonemik komaya neden olmaktadır.
Etiyoloji
Birincil rahatsızlık NAGS
geninde (17q21.31) oluşan mutasyonlar sonucu ortaya çıkmaktadır ve kısmı NAGS
aktivitesi eksikliğine neden olmaktadır. NAGS’ın ürünü NAG (N-acetylglutamate),
üreojenezin birinci basamağını katalize eden enzim olan korbomil fosfat sentez I’in
(CPSI) allosterik aktivatörüdür. N-Acetylglutamate
Sentaz Eksiliği, organik asitik hastalıklar, yağ asidi metabolizmasında oluşan
bozukluklar veya valproik asit tedavisi sonucunda da oluşabilmektedir.
Genetik Yatkınlık
N-Acetylglutamate
Sentaz Eksiliği’ne, 17q21 kromozomunda bulunan N-acetylglutamate sentaz geninin
mutasyona uğraması neden olmaktadır. Bu hastalık nesilden nesile otozomal
çekinik gen ile aktarılmaktadır. Bu yüzden kişinin bu hastalığa sahip olabilmesi için her
hücredeki sorumlu geninin ikisinin de mutasyona uğramış olması gerekmektedir.
Hasta bireyler, mutasyona uğramış bir geni anneden bir diğer geni de babadan
almaktadır. Bu durumda ebeveynler taşıyıcı olarak adlandırılmaktadır.
Taşıyıcılar otozomal çekinik gene sahip oldukları için bu hastalığın belirti
veya semptomlarını göstermezler. İki otozomal çekinik gene sahip bireyin çocuğu
olduğunda, bu çocuk %25 ihtimalle hasta, %50 ihtimalle taşıyıcı ve %25
ihtimalle ne hasta ne de taşıyıcı olacaktır.
Belirti ve Semptomları
N-Acetylglutamate
Sentaz Eksiliği’ne sahip hastaların %30-%79’unda çocuklukta görülen kas
hipotonisi, mide bulantısı, kusmaya rastlanmakta olup; %5-%29’unda da akut
hiperamonyemi ve gergin ruh haline rastlanmaktadır. Ayrıca hasta bireylerde
koma durumu, öğrenme güçlüğü, zihin bulanıklığı, kordinasyon eksikliği ve gelişimsel
gecikme görülebilmektedir. Bu semptomlar kişiden kişiye göre farklılık
göstermektedir ve kişi bütün semptomlara sahip olmak zorunda değildir. Semptomları
ve belirtileri ileri yaşlarda göstermeye başlayan insanlar, erken yaşlarda
gösterenlere veya besin tüketiminde proteine fazlaca yer veren insanlara göre
belirti ve semptomları daha hafif yaşarlar. Yenidoğanlarda N-Acetylglutamate
Sentaz Eksiliği’nin semptomları enerji eksikliği, yemek yemede isteksizlik,
nöbet, anormal vücut hareketleri, çok düzensiz ve zor nefes alma, değişken vücut
ısısıdır.
Teşhis Konulması
Nadir
görülen bir genetik hastalığın tanısının konması zor olduğu için doktorlar
genellikle kişinin tıbbi geçmişini, şüphe duyulan semptomlarını incelemekte;
fiziksel muayne ve laboratuvar testi sonuçlarının değerlendirilmesiyle de hastalık
teşhisini koymaktadır. Ayrıca hastalık karaciğerde azalış gösteren NAGS
aktivitesinden de anlaşılabileceği için teşhis DNA analizi ile de desteklenebilmektedir.
Hastalığın ana ayırıcı tanısı karbomil fosfat sentezi eksikliğidir.
Tedavi Edilmesi
N-Acetylglutamate
Sentaz Eksiliği hastaları için uygulanan ana tedavi günlük dozlarla karglumik
asit almaktır. Karglumik asit yapısal bakımdan NAGS’a benzemektedir ve korbomil fosfat sentez I’i (CPSI) aktive etmektedir.
Diğer
İsimleri
N-Acetylglutamate
Sentaz Eksiliği’ne Bağlı Hiperamonyemi
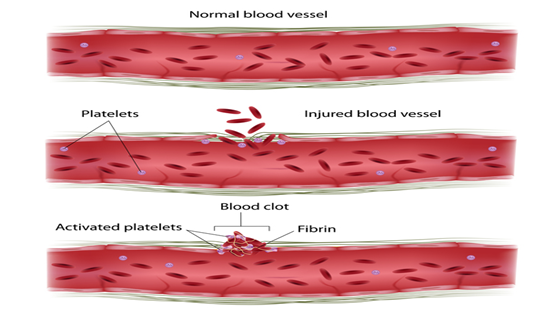
Faktör XI eksikliği ,
kanın pıhtılaşmasında rol alan faktör XI proteininin bir kıtlığına
(eksikliğine) bağlı olarak anormal kanamaya neden olabilecek bir hastalıktır. Bu durum, faktör XI proteininin eksiklik derecesine
bağlı olarak kısmi veya şiddetli olarak sınıflandırılır. Bununla birlikte,
protein eksikliğinin ciddiyetine bakılmaksızın, çoğu etkilenen birey göreceli
olarak hafif kanama problemlerine sahiptir.Faktör XI sendromuna sahip
kişilerdeki belirtiler aynı aile içerisinde bile değişebilir.Diğer genetik ve
çevresel faktörler hastalığın derecesinin belirlenmesinde etkilidir.
Belirti ve Semptomlar
Faktör XI eksikliği vakalarının
çoğu , faktör XI proteini yapmak için talimatlar veren F11 genindeki mutasyonlardan
kaynaklanır . Bu protein, kan pıhtılarını oluşturan
bir dizi kimyasal reaksiyon olan pıhtılaşma kaskadında rol oynar.Yaralanmaya
yanıt olarak. Bir yaralanmadan sonra pıhtılar kanamayı durdurmak ve kan
damarı onarımını tetiklemek için kan damarlarını kapatır.
F11 genindeki mutasyonlar ,
fonksiyonel faktör XI’nin eksikliğine (eksikliğine) neden olur. Bu
eksiklik pıhtılaşma kademesini bozar, kanın pıhtılaşma sürecini yavaşlatır ve
bu hastalıkla ilişkili kanama sorunlarına yol açar. Kalan fonksiyonel
faktör XI miktarı, özel mutasyona ve F11 geninin bir
veya her iki kopyasının, her hücrede mutasyona sahip olup olmamasına
bağlı olarak değişir . Bununla birlikte, etkilenen bireylerde kanama
problemlerinin ciddiyeti mutlaka kan dolaşımındaki faktör XI miktarına karşılık
gelmez ve aynı aile içinde bile
değişebilir. Diğer genetik ve çevresel faktörler muhtemelen bu
durumun ciddiyetinin belirlenmesinde rol oynar.
Faktör XI eksikliğinin en
sık görülen özelliği , özellikle ağız ve burun içini (travma veya burun boşlukları dahil) travma
veya ameliyat sonrası uzun süreli kanamadır.) veya idrar yolu. Kanama ameliyattan sonra tedavi
edilmezse, cerrahi alanda konjuge kandan (hematom) oluşan katı şişlikler
gelişebilir.
Bu hastalığın diğer belirti ve semptomları sık burun
kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını
içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet
kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip
olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri),
gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın
olmayan faktör XI eksikliği onlar
ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında
uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle
bu durumda meydana gelmez.
Bu hastalığın diğer belirti ve semptomları sık burun
kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını
içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet
kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip
olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri),
gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın
olmayan faktör XI eksikliği onlar
ağır etkilenen bireylerde oluşabilir
rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden
olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez.
Aşağıda bu hastalığı olan kişilerin sahip olabileceği
belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye
değişecektir. Aynı hastalığı olan insanlar listelenen tüm belirtilere
sahip olmayabilir. Bu tablo düzenli olarak İnsan Fenotip Ontoloji
((Human Phenotype Ontology [HPO]) isimli veri tabanı tarafından
güncellenmektedir. ([https://hpo.jax.org/)
Faktör
XI eksikliğinin dünya genelinde yaklaşık 1 milyon kişiden
1’ini etkilediği tahmin edilmektedir. Orta ve doğu Avrupa (Aşkenazi)
Yahudi soyuna sahip kişilerde, bu popülasyondaki 450 kişiden 1’inde meydana
gelen şiddetli bozukluk bozukluğu çok daha yaygındır. Araştırmacılar, faktör XI eksikliğinin gerçek
prevalansının bildirilenden daha yüksek olabileceğini öne
sürüyorlar , çünkü hastalığın hafif vakaları çoğu zaman tıbbi yardıma
gelmiyor.
Kalıtım Paterni /Deseni
Otozomal
resesif paternde ciddi faktör XI eksikliği azaldıbu, her bir hücrede F11 geninin
her iki kopyasının da mutasyonlara sahip olduğu anlamına
gelir . Bu bireylerin ebeveynlerinin her biri, mutasyona uğramış
genin bir kopyasını taşır ve kısmi faktör XI
eksikliğine sahiptir ; nadiren durumun ciddi belirtileri ve
semptomlarını gösterirler.
Bazı ailelerde bu durum otozomal dominant paternde kalıtsaldırbu, her
hücrede değiştirilmiş F11 geninin bir
kopyasının , bozukluğa neden olmak için yeterli olduğu anlamına
gelir . Bu gibi durumlarda, etkilenen bir kişinin durumu olan bir
ebeveyni vardır.
Kazanılan faktör XI eksikliği formu kalıtsal
değildir ve ailelerde çalışmaz.
Faktör XI eksikliği
sendromunun tanısı için 3 önemli belirti şu şekildedir:
1)Ağız ve burun içi travma veya ameliyat sonrası uzun
süreli kanama.
2)Sık sık burun kanaması, kolay morarma, deri altında
kanama ve diş etlerinin kanaması.
3)Bu hastalığa sahip olan kadınlarda ağır veya uzun
süreli adet kanaması (menoraji) ve doğumdan sonra uzun süreli kanama.
Ayrıca faktör XI eksikliği sendromu için bazı testler
yapılmaktadır. Ancak hastalık tanısı için gerekli değildir.
1)Genetik Test
Genetik test, kromozom, gen veya proteinlerdeki değişiklikleri tanımlayan
bir tıbbi test türüdür. Genetik testin sonuçları, şüpheli bir genetik
durumu onaylayabilir veya ekarte edebilir veya bir kişinin genetik hastalık
geliştirme veya geçme şansını belirlemeye yardımcı olabilir. Şu anda
1000’den fazla genetik test kullanılıyor ve daha fazlası geliştiriliyor.
Genetik test için çeşitli yöntemler kullanılabilir:
Moleküler genetik testler (veya
gen testleri), genetik bir bozukluğa yol açan varyasyonları veya
mutasyonları tanımlamak için tekli genleri veya kısa DNA uzunluklarını
inceler.
Kromozomal genetik testler,
genetik bir duruma neden olan bir kromozomun ekstra bir kopyası gibi büyük
genetik değişikliklerin olup olmadığını görmek için bütün kromozomları
veya uzun DNA uzunluklarını analiz eder.
Biyokimyasal genetik testler,
proteinlerin miktarını veya aktivite seviyesini inceler; herhangi
birindeki anormallikler, genetik bir bozuklukla sonuçlanan DNA’daki
değişiklikleri gösterebilir.
Genetik test isteğe bağlıdır. Testin sınırlamalar ve risklerin yanı
sıra faydaları olduğu için, test edilip edilmeyeceği konusundaki karar kişisel
ve karmaşık bir karardır. Bir genetikçi veya genetik danışman, testin
artıları ve eksileri hakkında bilgi vererek ve testin sosyal ve duygusal
yönlerini tartışarak yardımcı olabilir.
2)Kısmi Tromboplastin Zamanı(PTT):
Kısmi tromboplastin zamanı (PTT) kanın pıhtılaşmasının ne
kadar sürdüğünü gösteren bir kan testidir. Kanama probleminiz olup
olmadığını veya kanınızın pıhtılaşmaması durumunda size yardımcı olabilir.
3)Protrombin Zamanı(PT):
İlgili bir kan testidir.
Hastalığın Diğer İsimleri
F11 eksikliği
faktör 11 eksikliği
hemofili C
hemofili
C
plazma
tromboplastin öncül eksikliği
PTA
eksikliği
Rosenthal
faktörü eksikliği
Rosenthal
sendromu
Rosenthal
hastalığı
Faktör XI Eksikliği İçin Yurt Dışındaki Kuruluşlar


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}