Genel Bilgi
Okülo-aurikülo-vertebral spektrum (OAVS), birçok klinisyenin birbiriyle yakından ilişkili olduğuna inandığı ve aynı hastalığın ciddiyet aralığını temsil eden üç nadir hastalığa karşılık gelir. Bu bozukluklar doğumda belirgindir (doğuştan). Adından da anlaşılacağı gibi, gözler, kulaklar ve omurganın malformasyonlarını içerir.
Okülo-Aurikülo-Vertebral Spektrum (OAVS) Alt Bölümleri
- Goldenhar Sendromu
- Hemifakiyal Mikrozomi (HFM)
- Okülo-Aurikülo-Vertebral Bozukluk
Okülo-aurikülo-vertebral bozukluk (OAVD) hastalığın en hafif formunu temsil ederken, Goldenhar sendromu en şiddetli form olarak ortaya çıkar. Hemifakiyal mikrostomi orta bir form gibi görünmektedir.
Bozukluk, durumdan duruma değişiklik gösterebilen çok çeşitli belirtiler ve fiziksel özellikler ile tanımlanır. Bununla birlikte, bu tür anormallikler elmacık kemikleri, çene, ağız, kulaklar, gözler ve / veya omurganın (omurlar) kemiklerini tutma eğilimindedir. Çoğu durumda (yaklaşık% 60), bu tür malformasyonlar vücudun bir tarafını (tek taraflı) etkilese de, etkilenen bireylerin yaklaşık yüzde 10 ila 33’ü vücudun her iki tarafında bu tür malformasyonlara sahiptir (iki taraf), bir taraf tipik olarak daha fazla etkilenir diğerinden (asimetri). Bu gibi vakaların çoğunda, sağ taraf soldan daha ciddi bir şekilde etkilenir.
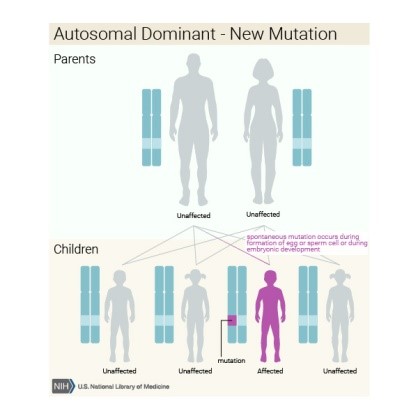
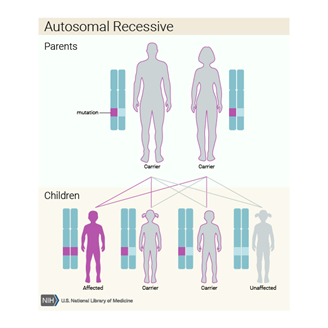
Çoğu durumda, OAVS rastlantısal olarak ortaya çıkar, görünür bir nedeni yoktur (sporadik). Bununla birlikte, bazı durumlarda, aile öyküleri otozomal dominant veya resesif kalıtım gösterir. Ek olarak, bazı araştırmacılar bu bozukluğa, muhtemelen çevresel faktörlerle (çok faktörlü kalıtım) kombinasyon halinde birçok genin etkileşiminden kaynaklanabileceğini öne sürmektedir.
Farklı görsellere ulaşabileceğiniz web sayfası: https://www.nature.com/articles/5201770
Genetik Değişiklikler / Etken Faktörler
Çoğu durumda, okülo-aurikülo-vertebral spektrum belirgin bir neden olmadan (sporadik) rastgele ortaya çıkar. Bununla birlikte, bazı durumlarda, otozomal dominant veya daha az sıklıkla otozomal resesif kalıtım öneren pozitif aile öyküleri tespit edilmiştir. Ek olarak, birçok araştırmacı, OAVS’nin, muhtemelen çevresel faktörlerle (çok faktörlü kalıtım) kombinasyon halinde birçok genin etkileşiminden kaynaklanabileceğini öne sürmektedir.
Açıklanamayan nedenlerden dolayı, hamilelik sırasında belirli ilaçlara (örneğin, retinoik asitli bazı akne ilaçları) ya da koşullara (örneğin diyabet) maruz kalmış kadınların, OAVS’ın özellik anormallikleri olan çocuklar olduğu anlaşılmaktadır. Ek olarak, OAVS ile ilgili ayırt edici özellikler de çeşitli kromozomal bozukluklarla birlikte ortaya çıkmıştır.
Belirti ve Semptomlar
Okülo-aurikülo-vertebral spektrum doğumda belirgin olan üç nadir bozukluğu temsil eder (konjenital) ve vakadan vakaya büyük ölçüde değişiklik gösterebilen geniş bir semptom yelpazesi ve fiziksel özellikler ile karakterize edilir. Bununla birlikte, bu tür anormallikler elmacık kemiklerini, çeneleri, ağzı, kulakları, gözleri ve / veya omurganın (omur) kemiklerini tutma eğilimindedir. Vakaların yaklaşık yüzde 60’ında, bu tür malformasyonlar vücudun bir tarafını (tek taraflı) içerir. Ancak, etkilenen bireylerin yaklaşık yüzde 10 ila 33’ünde, vücudun her iki tarafı da tutulabilir (iki taraflı), bir taraf diğerinden daha fazla etkilenir (asimetri). Bu gibi birçok durumda, sağ taraf soldan daha ciddi şekilde etkilenir.
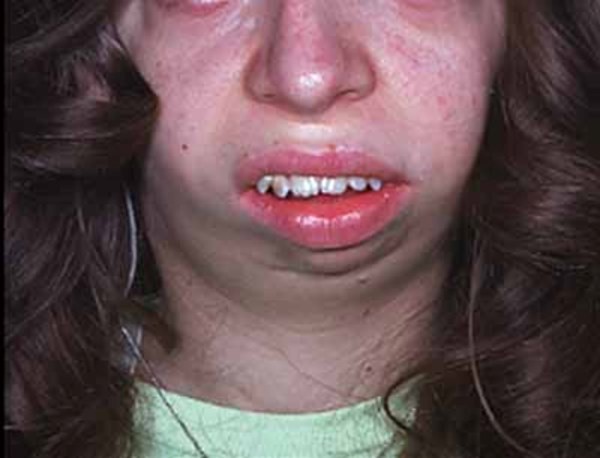
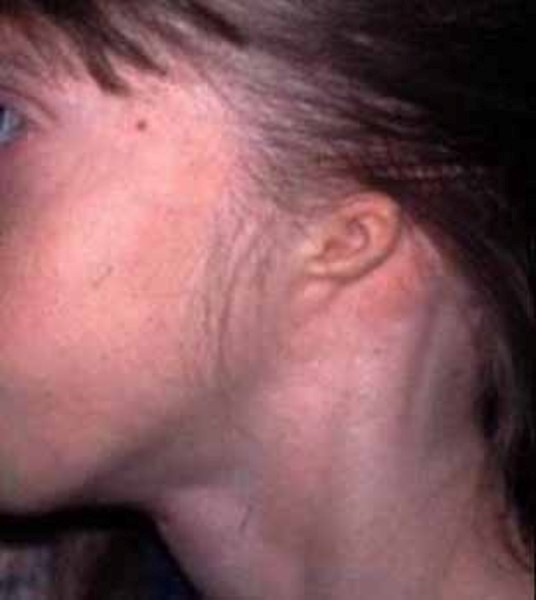
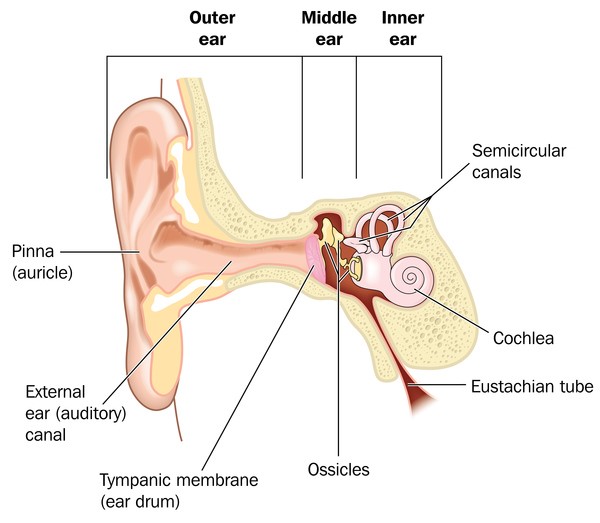
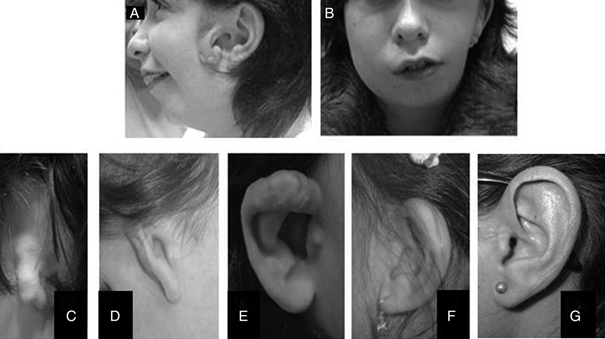
Bilinmeyen nedenlerden dolayı hemifasiyal mikrozomi (HFM) yüzün sadece sağ tarafını etkileme eğilimindedir. HFM’de, hem çene hem de göz etkilenen tarafta büyük ölçüde daha küçük olabilir. Etkilenen taraftaki yanak, o taraftaki elmacık kemiklerinin gelişmesi nedeniyle daha düz görünebilir. Dış kulak daha küçük (mikrotia) veya hatta yok (anotia) olabilir. İşitme kaybı da olabilir. İstihbarat etkilenmez.
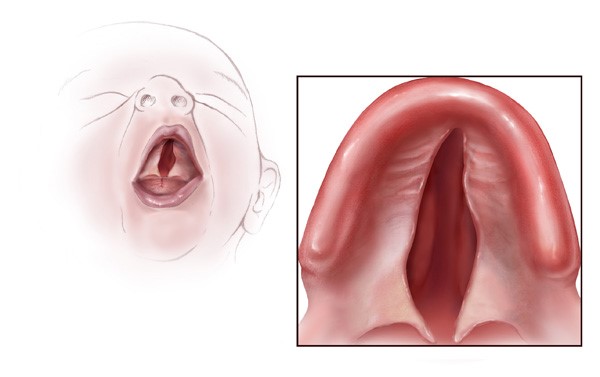
OAVS’nin Goldenhar varyantına sahip kişiler, çoğu HFM belirtisinin tümü olmasa da ortaya çıkar, ancak vakaların yüzde 10 ila 33’ünde, semptomlar yüzün her iki tarafını da etkiler (iki taraflı). Yarık dudak ve / veya yarık damak mevcut olabilir, ancak tek başına yarık damak varlığı daha yaygındır. Dilin ve yanakların kasları konuşmada ciddi zorluklara neden olabilir. Gözün bazı dokuları kapanmayabilir ve değişken büyüklükte bir çentik (koloboma) şeklinde ortaya çıkabilir. Vakaların yaklaşık üçte birinde, hasta gözde bir kist (dermoid kist) ile kendini gösterir. Ayrıca, Goldenhar sendromlu hastalar, kalp defekti ve böbrek problemleri ile birlikte ortaya çıkabilir. Goldenhar sendromlu kişiler bir tarafta az gelişmiş böbreklere ve hatta etkilenen tarafta böbrek eksikliğine sahip olabilir. İki veya daha fazla omur birbirine kaynaşmış veya örülmüş olabilir.
Genetik Görülme Sıklığı
OAVS, erkekleri kadınlardan yaklaşık 3: 2 oranında daha sık etkiler. Tıp literatüründe bozukluğun ortaya çıkma oranı ile ilgili bazı anlaşmazlıklar vardır. Rapor edilen tahminler 3000 ila 5000 canlı doğumdan birinden, 25.000-40.000 canlı doğumdan birine kadar değişmektedir. OAVS ile ilişkili fiziksel özelliklerin çoğu, doğumda (doğuştan) belirgindir; çoğu durumda yaklaşık dört yaşına kadar belirgin olmayabilecek olası yüz asimetrisi dışındadır.
Kalıtım Paterni /Deseni
Okülo-aurikülo-vertebral spektrum (OMIM164210) kulağın anomalileri (çoğunlukla mikrotia), hemifasiyal mikrozomi ve vertebral kolonun defektleri ile karakterize fenotipik ve muhtemelen genetik olarak heterojen bir hastalıktır. İlişkili klinik bulgular göz ve beyin anomalilerini ve gelişimsel gecikmeyi içerir.
| Kraniyofasiyal Mikrozomi Genel Bakış: Dahil Olan Fenotipler |
| Hemifakiyal mikrozomi Okülo-aurikülo-vertebral spektrum Goldenhar sendromu Birinci ve ikinci branşsal ark sendromu Otomandibular disostoz Facio-auriculo-vertebral sendrom Lateral yüz displazisi |
Kraniyofasiyal mikrozomi (CFM), öncelikle birinci ve ikinci dal kemerlerinden türetilen yapıları içeren bir tür yanlış biçimlendirme spektrumunu içerir. Karakteristik bulgular maksiller ve / veya mandibular hipoplaziden kaynaklanan yüz asimetrisini; preauriküler veya yüz etiketleri; mikrotiya (dış kulağın hipoplazisi), anotia (dış kulak yokluğu) veya aural atreziyi (dış kulak kanalının yokluğu) içerebilen kulak malformasyonları; ve işitme kaybı. Ciddiyet, normal görünen bir kulağın önünde küçük bir cilt etiketi bulunan ince yüz asimetrisinden, iki taraflı tutuluma (tipik olarak asimetrik), kulak kanallarının atrezili mikrorotiyal / anotiye, mikroftalmi ve ağır mandibular hipoplaziden solunum yetmezliğine kadar değişebilir. Yarık dudak ve / veya damak dahil diğer kraniyofasiyal malformasyonlar görülebilir.
CFM en sık bilinmeyen etiyolojiye sahip tek taraflı bir vaka (yani, ailedeki tek bir bireyde ortaya çıkma) olarak ortaya çıkar ; tekrarlama riskleri ampiriktir. CFM’li bir kişinin kalıtsal veya de novo kromozom anomalisine sahip olduğu tespit edilirse , bu durum için genetik danışma belirtilir. Bazen otozomal dominant veya otozomal resesif kalıtım görülür. Bir proband CFM’ye sahipse ve CFM ailesinde rapor edilmemiş bir aile öyküsü yoksa, sibs riski% 2 -% 3’dür, ancak bu düşük penetrasyon ve bazı ince özellikler için doğru aile öyküsü elde etmenin zorluğu nedeniyle hafife alınabilir .
Teşhis Yöntemleri ve Tedavileri
Teşhis
Nadiren, okülo-aurikülo-vertebral spektrum doğumdan önce (doğum öncesi) ultrason görüntüleme gibi özel testlerle tespit edilebilir. Fetal ultrasonografide, yansıyan ses dalgaları, gelişmekte olan fetüsün bir görüntüsünü oluşturmak için karakteristik bulguları ortaya çıkarmak için kullanılabilir. OAVS durumunda, bu bulgular alt çenede (mandibula) kemik varlığına veya yokluğuna, dış kulaklarda ciddi anormalliklere, yarık damakta ve / veya yarık dudağa bağlıdır.
OAVS ayrıca doğumdan sonra (doğum sonrası) ayrıntılı bir klinik değerlendirme, karakteristik fiziksel bulguların belirlenmesi ve ileri görüntüleme teknikleri ile teşhis edilebilir ve / veya doğrulanabilir.
Potansiyel olarak okulo-aurikülo-vertebral spektrum bozuklukları ile ilişkili spesifik anormallikleri doğrulamak için çeşitli uzmanlık testleri yapılabilir. Örneğin, bilgisayar destekli tomografi (BT) taraması, işitme kaybına katkıda bulunabilecek orta kulak anormalliklerinin tespitinde önemli bir yardımcı olabilir. Gelişmiş görüntüleme teknikleri ayrıca kafatasının, omurganın, akciğerlerin ve / veya böbreklerin diğer potansiyel anormalliklerini tespit etmek ve / veya onaylamakta yardımcı olabilir. Bazı durumlarda, hastalıkla ilişkili olabilecek konjenital kalp kusurlarının varlığını tespit etmek ve / veya doğrulamak için ek özel testler (örneğin, ekokardiyogramlar, elektrokardiyogramlar, kalp kateterizasyonu, özel röntgen çalışmaları vb.) yapılabilir. .
Mikroftalmi veya anoftalmi, epibulbar dermoidler ve lipodermoidler, şaşılık, vb. Gibi bazı göz (oküler) anormallikleri tespit etmek, doğrulamak ve / veya karakterize etmek için gözün içini görselleştiren bir aletle (opthalmoscope) inceleme yapılabilir.
OAVS’li yenidoğanlarda yutma ve beslenme zorlukları, özofagus atrezisi ve trakeoözofageal fistül gibi anormallikler gösterebilir. Bu anormallikler, sıvıyı vücuda enjekte etmek veya sıvıyı vücuttan boşaltmak için kullanılan esnek, içi boş bir tüp aracılığıyla tespit edilebilir (kateter). Ağızdan mideye geçemezse konjenital malformasyonlar mevcut olabilir.
Tedavi
OAVS tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, tedaviye kapsamlı ve sistematik bir yaklaşım sağlamak için birlikte çalışması gerekebilecek bir uzmanlar ekibinin koordine çabalarını gerektirebilir. Bu uzmanlar çocuk doktorlarını içerebilir; kulak, burun ve boğaz rahatsızlıklarını teşhis ve tedavi eden doktorlar (kulak burun boğaz uzmanları); göz uzmanları (göz doktorları); nöroloji; kalp (kardiyologlar) ve / veya akciğer (kardiyotorasik) cerrahları; böbrek (nefrologlar), idrar yolu (ürologlar) ve sindirim sistemi (gastroenterologlar) rahatsızlıklarının tanı ve tedavisinde uzmanlaşmış doktorlar; estetik cerrahlar; işitme problemlerini değerlendiren ve tedavi eden uzmanlar (odyologlar); konuşma patologları; ve / veya diğer sağlık profesyonelleri.
- Bu hastalarda genellikle rekonstrüktif cerrahi gereklidir.
- Mandibular hipoplazisi olanlar, kaburga kemiği greftleri kullanılarak rekonstrüksiyonlara sunulabilir ve az gelişmiş bir maksilla, kemik distraksiyonu ve osteogenez ile uzatılabilir.
- Dış kulağın rekonstrüktif ameliyatları 6 ila 8 yaşlarında yapılabilir.
- Daha hafif tutulumlu hastalarda ergenlik döneminde rekonstrüktif çene ameliyatları yapılabilir.
- Bu sendromlu çocuklar psikososyal zorluklar açısından daha yüksek bir risk taşır, bu nedenle hastalara ve ailelere destek önerilir.
- Prognoz, sistemik tutulumu olmayan komplike olmayan vakalarda genellikle iyidir; Bununla birlikte, bazı ciddi vakalarda doğumdan itibaren erken cerrahi müdahale gerekebilir.
- Bu bölgelerdeki doğum kusurları riskindeki artış nedeniyle böbrek ve kalp ultrasonları önerilebilir.
Hastalıkla İlişkili Genler
Aşağıdaki bozuklukların belirtileri okülo-aurikülo-vertebral spektrumun belirtilerine benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Treacher Collins sendromu, kafatasının belirli kısımlarının (örneğin, supraorbital jantlar ve elmopatik kemerler) ve alt çenenin az gelişmesine (hipoplazisi) bağlı olarak kranyofasiyal bölgenin belirgin anormallikleri ile karakterize oldukça nadir bir genetik hastalıktır. Treacher Collins sendromu ile ilişkili semptomlar ve fiziksel özellikler vakadan duruma ciddi olarak değişebilse de, kraniyofasiyal anormallikler elmacık kemiklerini, çeneleri, ağzı, kulakları ve / veya gözleri tutma eğilimindedir. Bu tür kranyofasiyal malformasyonlar az gelişmiş (hipoplastik) veya eksik elmacık kemikleri; tamamiyle gelişmiş, anormal derecede küçük bir alt çene (mandibular hipoplazi ve mikrognati); alışılmadık derecede büyük bir ağız (makrostomi); ağız çatısının malformasyonları (damak); ve / veya yanlış hizalanmış dişler (maloklüzyon) gibi diş anormallikleri. Etkilenen bebekler ayrıca az gelişmiş (hipoplastik) ve / veya kör biçimli veya kulak kepçeli dış kulak kanallarına (atreziye) sahip olmayan (mikro) kulaklarda dışa dönük kulaklara sahip olabilirler (işitme bozukluğu). Ek olarak, bozukluğu olan bebekler aşağı doğru eğimli göz kapağı kıvrımlarına (palpebral fissüler), alt göz kapaklarının dış üçte birinden kısmen veya tamamen doku yokluğuna (colobomas) ve / veya ilave göz anormallikleri gösterebilir. Vakaların yaklaşık yüzde 40’ında, Treacher Collins sendromu otozomal dominant kalıtıma sahiptir. Bununla birlikte, vakaların yaklaşık yüzde 60’ında pozitif bir aile öyküsü bulunamamıştır. Araştırmalar, bu gibi vakaların rastgele ortaya çıkan, belirgin bir neden olmadan (sporadik) yeni genetik değişiklikleri (mutasyonları) temsil ettiğini göstermektedir. (Bu hastalık hakkında daha fazla bilgi için,
Erken fetal gelişim sırasında birkaç kusurdan kaynaklanan nadir bir hastalık olan CHARGE birlikteliği, vücudun birkaç organ sistemini etkileyen anormallikler ile karakterizedir. CHARGE (C) gözün, özellikle gözün renkli kısmının (iris) olobomasını temsil eden, iris’e anormal bir “anahtar deliği” şekli veren bir kısaltmadır; (H) Fallot Tetralojisi, ventriküler ve / veya atriyal septal defektler ve patent duktus arteriosus dahil olmak üzere toprak defektleri; (A) burun arkasını boğaza bağlayan geçitlerin veya koana’nın daraltılması veya tıkanması, yani normal burun nefesinin önlenmesi; (R) bazı durumlarda zihinsel ve psikomotor geriliğin yanı sıra, büyüme ve gelişmenin durması; (G) enital ve idrar anomalileri; ve (E) dış kulakların ve orta kulakların kemiklerinin bozulmaması, östaki tüplerinin yanlış çalışması, kulak kanallarının tıkanması ve / veya işitme kaybı gibi anormallikler. CHARGE ilişkisinin tanısını doğrulamak için bu karakteristik bulgulardan dördü mevcut olmalıdır. Klasik özelliklere ek olarak, CHARGE Association ile bireyler ayrıca kranyofasiyal anormallikler, böbrek ve merkezi sinir sistemi malformasyonları ve / veya trakeoözofageal fistül ve / veya deliksiz anüs gibi diğer anormallikler de gösterebilirler. CHARGE ilişkisinin kesin nedeni bilinmemektedir; Bununla birlikte, çoğu vakanın rastlantısal olarak ortaya çıktığı ve belirgin bir sebep olmadığı düşünülmektedir (sporadik).
Fetal gelişim kusurlarından kaynaklanan nadir bir hastalık olan VACTERL birleşmesi, vücudun birkaç organ sistemini etkileyen konjenital anormallikler ile karakterizedir. VACTERL (V) hemivertebra ve alt vertebra (sakrum) malformasyonu dahil olmak üzere ertebral anormallikleri temsil eden bir kısaltmadır; (A) nal atrezi, anal açılmasının olmadığı bir durum; (C) arter defektleri, özellikle ventriküler septal defektleri; (T) rakeo (E) sifageal fistül; (R) böbrek ve hidronefroz yokluğu dahil enal anormallikler; ve ön kol kemiklerinden birinin (radyal displazi) ve diğer (L) imb defektlerinin yanlış gelişimi. Semptomlar çeşitli kombinasyonlarda ortaya çıkabilir ve birçok bilinen bozukluğun tezahürü olabilir. Etkilenen bireyler ayrıca vücudun diğer sistemlerini içeren ek anormallikler de gösterebilirler. Çoğu durumda, VACTERL ilişkisinin, rastgele bir nedenden ötürü rastgele meydana geldiği düşünülmektedir (sporadik); Bununla birlikte, araştırmacılar bazı vakaların X’e bağlı veya otozomal resesif genetik bir özellik olarak kalıtımsal olabileceğini öne sürmektedir.
Townes-Brocks sendromu, doğumda görülen ve doğuştan görülen, nadir görülen kalıtsal bir hastalıktır. Hastalıkla ilişkili semptomlar ve fiziksel özellikler vakadan duruma geniş ölçüde değişebilse de, anormallikler yüz, kulaklar, kollar ve bacaklar (uzuvlar), gastrointestinal sistem ve böbrekleri etkileme eğilimindedir. Bozukluğu olan kişilerde, yüzün bir tarafı diğerinden daha küçük görünebilir (hemifasiyal mikrozomi). Kulak anormallikleri, dış kulakların yanlış şekillenmesini, aşırı cilt etiketlerini ve / veya kulakların önündeki girintileri (preauriküler etiketler ve / veya çukurlar) ve / veya iç kulağın anormallikleri (duyusal işitme kaybı) nedeniyle duyma bozukluğunu içerebilir. Etkilenen bireyler ayrıca baş parmaklarında, ekstra parmaklarda (polidaktili), iki veya daha fazla parmak ve / veya ayak parmağında (eş zamanlı olarak) bağlanma biçiminde bozukluklara sahip olabilir. ve / veya diğer uzuv düzensizlikleri. Ek olarak, Townes-Brocks sendromlu bireyler anal açıklığın yokluğunu gösterebilir (deliksiz anüs); rektum ve cinsel organlar arasında anormal geçişler (rektovajinal veya rektoperineal fistül); az gelişmiş böbrekler (böbrek hipoplazisi); idrarın mesaneden geriye bir üretere (vezikoüreteral reflü) aktığı bir durum; ve / veya diğer ilgili anormallikler. Ek olarak, bazı durumlarda, etkilenen bireylerde kalp ve üreme organlarında anormallikler de olabilir. Townes-Brocks sendromu otozomal dominant kalıtıma sahiptir.
Branchio-oto-renal (BOR) sendromu, öncelikle kulakları, boynu ve boğazı ve böbrekleri etkileyen anormalliklerle karakterize nadir görülen kalıtsal bir hastalıktır. Etkilenen bireyler kulakların önünde aşırı cilt etiketleri (preauriküler etiketler), orta ve iç kulağın malformasyonu, hatalı biçimlendirilmiş dış kulaklar ve hafif ila şiddetli iletken ve / veya duyusal işitme kaybı gösterebilir. Ek anormallikler, boğazdan boynun dış yüzeyine (dal fistülü); bademcik bölgesinde anormal bir açıklık, kist veya kitle; gözyaşı kanallarının (lakrimal kanalların) daralması (darlığı) ve / veya yokluğu (aplazisi); ve / veya uzun, dar bir yüz, ağız çatısının eksik kapanması (yarık damak), derin bir overbite ve / veya yüzün belirli kaslarının felci dahil kraniofasiyal anormallikler. BOR sendromu olan kişilerde, olağandışı şekilli böbrekler, böbreklerin toplama sisteminin çoğaltılması ve / veya böbreklerin az gelişmişliği (hipoplazi) dahil olmak üzere, genellikle hafif ila şiddetli böbrek (böbrek) anormallikleri vardır. branşio-oto-renal sendrom otozomal dominant genetik özellik olarak kalıtsaldır.
Hastalığın Diğer İsimleri
- OAV Spektrumu
- Okülo-Aurikülo-Vertebral Displazi
- Facio-Auriculo-Vertebral Spektrum
- FAV
- Birinci ve İkinci Branş Kemer Sendromu
Kaynaklar
- https://rarediseases.org/rare-diseases/oculo-auriculo-vertebral-spectrum/
- https://www.ncbi.nlm.nih.gov/pubmed/16378924
- http://www.ajnr.org/ajnr-case-collections-diagnosis/oculo-auriculo-vertebral-spectrumgoldenhar-syndrome
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=141132
- https://www.ncbi.nlm.nih.gov/books/NBK5199/