Aicardi Sendromu neredeyse sadece kadınlarda meydana gelen 3 özellikle karakterizedir.Aicardi Sendromlu insanlar beynin sağ ve sol yarısındaki bağlantı dokusunun az gelişmişliğine yada yokluğuna sahiptirler(corpus callosumun agenezi yada disgenezisi).Süt çocukluğunda başlayan nöbetleri vardır(infantil spazmlar) ki tekrarlayan nöbetlere(epilepsi) gelişme eğilimindedirler bunun tedavi edilmesi zordur.Etkilenmiş bireylerde ayrıca korioretinal laküna vardır ki gözün arkasındaki(retina) ışığa duyarlı dokuda kusurlardır.
Aicardi Sendromlu insanlarda sıklıkla ek olarak beyin anormalileri vardır,beynin iki tarafı arasında asimetri,beyin kıvrımlarının ve oluklarının boyut olarak küçük olması yada sayısının azalması,kistler ve beynin merkezine yakın sıvı dolu kavitelerin(ventrikül) büyümesi bunları içerir.Bazılarının alışılmadık şekilde küçük kafaları vardır(mikrosefali).Çoğu etkilenmiş bireyde ılımlıdan şiddetliye gelişimsel gerilik ve entelektüel disabilite vardır buna rağmen hastalığa sahip bazı insanlar ılımlı disabiliteye sahiptirler.
Korioretinal lakünaya ek olarak,Aicardi Sendromlu bireyler diğer göz anomalilerinden küçük yada zayıf gelişmiş gözler(mikroptalmi) gibi yada gözden beyine bilgi taşıyan yapıda yani optik sinirde boşluk veya deliğe(kolobom) sahip olabilirler.Bu göz anomalileri etkilenmiş bireylerde körlüğe sebep olurlar.
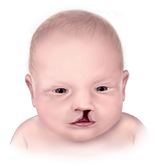
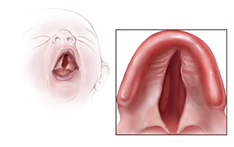
Aicardi Sendromlu bazı bireylerde alışılmamış yüz özellikleri vardır bunlar üst dudakla burun arasındaki alanın(filtrum) kısalığı,büyük ucu kalkık ve yassı burun,büyük kulaklar ve seyrek kaşları içerir.Bu durumun diğer özellikleri küçük eller,el malformasyonları ve omurganın progresif anormal ilerleyen eğriliğine(skolyoz) yönlendiren omurga ve kaburga anomalilerini içerirler.Genellikle konstipasyon yada diyare,gastroözofagal reflü ve beslenmekte zorluk gibi gastrointestinal problemleri vardır.
Aicardi Sendromunun şiddeti çeşitlilik gösterir.Hastalığa sahip bazı insanların çok şiddetli epilepsisi vardır ve çocukluğa kadar hayatta kalamayabilirler.Az şiddetli bireyler ılımlı işaret ve semptomlarla yetişkinliğe kadar yaşayabilirler.
Sebepleri
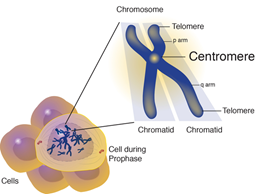
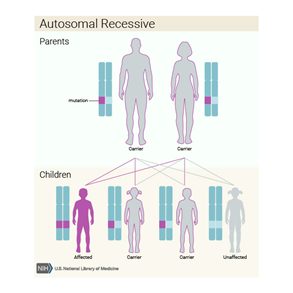
Aicardi Sendromu büyük ihtimalle X kromozomunda yerleşmiş gende yeni mutasyon sebebiyle olur.Aicardi Sendromuna sebep olan gen bilinmiyor.Raporda TEAD1 ve OCEL1 genlerinde değişim Aicardi Sendromlu 2 kızda tanımlanmıştır bu değişiklikler büyük gruptaki Aicardi Sendromlu diğer kızlarda doğrulanamamıştır.Böylelikle bu genler Aicardi Sendromuna sebep olarak görünmemektedir.Bu durumun erkeklerde ölümcül olduğu farz edilmektedir.
Aicardi Sendromlu kızın ailesi tipik olarak etkilenmemiştir.Etkilenmiş anneden çocuğuna Aicardi Sendromunun iletimi rapor edilmemiştir.Ayrıca diğer aile üyeleri de genellikle artmış risk altında değildirler.
Sıklık
Aicardi Sendromu çok nadir bir hastalıktır.ABD’de yaklaşık 105.000-167.000 yeni doğanda 1 meydana gelir.Araştırmacılar dünya çapında yaklaşık olarak 4.000 etkilenmiş birey olduğunu tahmin etmektedirler.
Bu Durumun Diğer İsimleri
- Aicardi’nin Sendromu
- Oküler anormaliler ve infantil spazmlarla birlikte corpus callosum agenezi
- Korioretinal anormaliyle birlikte corpus callosum agenezi
- Kalozal agenezi ve oküler anormali
- ACC ile birlikte korioretinal anormaliler
Aicardi Sendromunun Alt dalları
Alt dalları bulunmamaktadır.
İşaret ve Semptomlar
Aicardi Sendromu tipik olarak 4 ay ile 4 yıl arasında istemsiz kas spazmlarıyla başlar.Diğer semptomlar epilepsi,entelektüel disabilite,büyük kas güçsüzlüğü(hipotoni),anormal küçük kafa(mikrosefali) ve retina ile gözün arkasındaki sinirin(kolobom) gelişimini tamamlayamaması ve/yada kaburgalarda ve/yada spinal sütunda anomalileri içerir.Aicardi Sendromlu her yaştaki tüm çocuklarda motor gelişimde önemli ölçüde gecikme vardır.Üst solunum yolu enfeksiyonlarından dolayı çocukluk boyunca Aicardi Sendromu yaşamı tehdit edici olabilir.
Kalıtım Paterni
Bilinen Aicardi Sendromlu olguların neredeyse tamamı seyrektir,bu jenerasyon vasıtasıyla geçmemiştir ve aile hikayelerinde bu hastalığa sahip bireyler meydana gelmemiş demektir.Bu hastalığın yeni gen mutasyonları sonucu olduğuna inanılmaktadır.
Aicardi Sendromu X’e bağlı dominant olarak sınıflandırılır.Hastalıkla bağlantılı gen bilinmiyorken,X kromozomunda yerleşmiş olduğuna inanmaktadır.Kadınlarda(2 X kromozomuna sahiptirler),her hücredeki genin bir yada iki kopyasında mutasyon hastalığa sebep olması için yeterlidir.Erkekler(Sadece 1 X kromozomuna sahiptirler) her hücredeki genin bir kopyasındaki mutasyon neredeyse her zaman çok erken gelişimde ölümcüldür,bu yüzden Aicardi Sendromlu bebeklerin neredeyse tümü kızdır.Yinede,her hücrede bir tane ekstra X kromozomlu az sayıda etkilenmiş erkekler(47,XXY) tanımlanmıştır.47,XXY Kromozom paternli erkekler ayrıca Klinefelter Sendromu denen duruma da sahiptirler.
Standart Terapiler
Tedavi
İlaçlar Aicardi Sendromunun sebep olduğu nöbetleri baskılayabilir.Nöbetleri tedavi etmek genellikle zordur.Doktorun bir takım ilaçları deneyip hangisinin en iyi çalıştığını görmeye ihtiyacı vardır.Çalışmalar Aicardi Sendromlu herkes için çalışan tek bir ilaç olmadığını gösteriyor.
Kaynakça