Galloway mowat sendromu nadir ve nörodejeneratif bir hastalıktır. Bu hastalığa sahip kişilerde vücut gelişiminde ve fiziksel olarak anormallikler görülür. Bu hastalık ailedeki taşıyıcı genlerle ortaya çıkar. Yani hastalığa sahip olmayan ama taşıyıcı olan anne ve babanın çocuğunda bu hastalık görülebilir. Hasta kişilerin semptom ve belirtileri birbirinden farklı olabilir. Bu farklılıklara göre tedavi görürler.
Belirti ve Semptomlar
Semptom ve
belirtiler ; başın küçük olması (microcephaly) , gelişmede gerilik , nöbetler; nefrotik sendrom(böbrek hastalığı); hiatal
herni(mide fıtığı); optik atrofi(göz hastalığı) ; hareket bozuklukları; ve
zihinsel engellilik.
Genetik Görülme Sıklığı
Hastalık ilk olarak 1968de iki kardeşte görülen erken başlangıçlı nefrotik sendromu (böbrek hastalığı) , başın küçük olması (mikrosefali) ve hiatus hernisi ( mide fıtığı) ortaya çıktığından beri bu hastalığa dair yaklaşık 40 vaka bildirilmiştir.
Kalıtsal Patern
Hala tam
olarak belli olmasa da kalıtsal olarak aktırılması için iki tarafta da aynı
genin mutasyona uğramış ve kişilerin taşıyıcı(hastalık belirtisi göstermeyen)
olması gerekir. İki taşıyıcı insanın çocuklarının hastalığa sahip olma
olasılığı -otozomal olduğundan dolayı- %25, taşıyıcı olma olasılığı %50, hastalıktan hiç
etkilenmeme olasılığı ise %25tir.
Teşhis Yöntemleri ve Tedaviler
Galloway-Mowat sendromu doğumdan sonra ayrıntılı bir
klinik değerlendirme, karakteristik fiziksel bulgular, özel laboratuar
testleri, görüntüleme teknikleri ve genetik testlerle teşhis edilebilir.
Hastalığa
sahip olan çocuklar çoğunlukla birkaç yıldan fazla yaşamazlar. Tedavi ise
kişideki belirli semptomlara bağlı değişir. Mide , göz ve diğer organ
hastalıkları bir şekilde tedavi edilmeye çalışılsa da hastalığın genel olarak
kesin çözümlü bir tedavisi yok.
Hastalıkla İlişkili Genler
Bu hastalık WDR73 genindeki mutasyonla ortaya çıkmış
olabilir ya da kalıtsal olarak otozomal bir şekilde aktarılabiliyor olabilir ya
da başka bilinmeyen genler tarafından etkileniyor olabilir. Hala kesin olarak
bir gen bulunamamıştır.
L1 sendromu, öncelikle sinir sistemini etkileyen ve neredeyse sadece erkeklerde görülen bir grup durumu tanımlar. Bu koşullar şiddette değişiklik gösterir ve en ağırdan en azına kadar, Sylvius (HSAS) su kemeri stenozu, MASA sendromu, spastik parapleji tip 1 ve X’e bağlı komplike korpus kallusum agenezi olan X’e bağlı hidrosefali içerir.
HSAS, durumun karakteristik özelliklerinin
kısaltmasıdır: beyinde bir sıvı (hidrosefali) genellikle doğumdan önce ortaya
çıkan bir sıvı birikimi, kas sertliği (spastisite), avuç içlerine kalıcı olarak
bükülen baş parmaklar (uyarılmış başparmaklar) ve Beyindeki bir geçidin
daraltılması (darlığı) Sylvius’un su kemeri olarak adlandırılır. HSAS’lı
bireylerde Sylvius’un su kemeri stenozu, ventrikül adı verilen sıvı dolu
boşluklardan beyin omurilik sıvısının (BOS) akışını engelleyerek hidrosefali
oluşturur. HSAS’lı bireyler sıklıkla ciddi zihinsel engellidir ve nöbet
geçirebilirler.
MASA
sendromu ayrıca, hafif ila orta, gecikmeli konuşma (afazi), spastisite ve
eklenmiş başparmaklar arasında değişebilen zihinsel engelli (zihinsel gerilik)
durumun karakteristik özellikleri için de adlandırılmıştır. MASA sendromlu
bireylerde ventrikülde hafif genişleme olabilir.
Spastik
parapleji tip 1, ilerleyici kas sertliği (spastisite) ve uzuvların paralizi gelişimi
(parapleji) ile karakterizedir. Etkilenen bireyler ayrıca hafif ila orta
dereceli zihinsel engellidir. Spastik parapleji tip 1 olan kişilerde genellikle
beyin yapılarında önemli anormallikler yoktur.
X’e
bağlı komplike korpus kallosum agenezisi, beynin sol ve sağ yarısını (korpus
kallosum) bağlayan dokunun az gelişmişliği (hipoplazi) veya yokluğu (agenezisi)
ile tanımlanır. Bu durumu olan insanlar spastik paraplejiye sahip olabilir ve
hafif ila orta derecede zihinsel engelli olabilir.
L1 sendromlu
bireylerin yaşam beklentisi, belirti ve semptomların ciddiyetine bağlı olarak
değişir. Ciddi derecede etkilenen bireyler doğumdan sadece kısa bir süre daha hayatta
kalabilirken, hafif özellikleri olanlar yetişkinlikte yaşar.
L1
sendromunu oluşturan koşulların bir zamanlar farklı bozukluklar olduğu
düşünülüyordu, ancak genetik bir nedeni paylaştığı tespit edildiğinden, şimdi
aynı sendromun bir parçası olarak kabul ediliyorlar. Aynı mutasyonun neden
olduğu L1 sendromlu aile üyeleri, durumun farklı biçimlerine sahip olabilir.
Aşağıda (L1 Sendromlu bir hastada genel problemler) görsel kaynağı: https://www.sciencedirect.com/science/article/pii/S030384671830221X
Genetik Değişiklikler/ Etken Faktörler
L1 sendromuna L1CAM genindeki mutasyonlar neden olur.
L1CAM geni, sinir sistemi boyunca bulunan Ll hücre yapışma molekülü proteinini
(Ll proteinine kısaltılmış) üretmek için talimatlar sağlar. Bu protein,
hücrelerin birbirine yapışmasına yardımcı olmak için komşu nöronlardaki
proteinlere bağlandığı (bağlandığı) sinir hücrelerinin (nöronlar) yüzeyinde
bulunur. L1 proteini beyin gelişimine, düşünme yeteneğine, hafızasına ve
hareketine katkıda bulunan nöronların birçok işlevinde rol oynar.
L1 sendromuna neden olan L1CAM gen mutasyonları, hücre-hücre yapışmasını kolaylaştıramayan veya
çeşitli nöronal fonksiyonlara katılamayan bir L1 proteinine yol açar. Bu
fonksiyonların bozulması muhtemelen beynin büyümesini ve gelişmesini
engelleyerek L1 sendromunun belirti ve semptomlarına yol açar.
Bazı L1CAM gen mutasyonları, anormal derecede kısa ve işlevsel olmayan bir
proteinin üretilmesine veya proteinin tamamen yok olmasına neden olur. Bu
mutasyonlar tipik olarak, sıklıkla HSAS olmak üzere, ciddi L1 sendromu vakalarına
yol açar. Diğer mutasyonlar, proteinin yapısını değiştirir, proteinin hücre
yüzeyindeki diğer proteinlerle etkileşime girme kabiliyetini azaltır veya
proteinin ihtiyaç duyulduğu yerde hücre yüzeyine ulaşmasını önler. Bu
mutasyonlar tipik olarak daha az ciddi L1 sendromu vakalarına, genellikle MASA
sendromuna veya bu durumun diğer hafif formlarına neden olur.
Bir mutasyonun L1 proteini üzerindeki etkisi, bazen
durumun ciddiyeti hakkında bir ipucu sunsa da, aynı veya benzer mutasyonlara
sahip bireylerin çok farklı belirti ve semptomları olabilir.
Belirti ve Semptomlar
L1 sendromu, farklı derecelerde şiddet
derecesine sahip hidrosefali, bacakların spastisitesi ve uyarılmış başparmaklar
ile karakterize hafif-şiddetli konjenital X’e bağlı bir gelişimsel bozukluktur.
Sendrom, aşağıdakileri içeren bir hastalık spektrumunu temsil eder: Sylvius
(HSAS) su kemeri stenozuna sahip X’e bağlı hidrosefali, MASA sendromu, X’e
bağlı komplike kalıtsal spastik parapleji tip 1 ve X’e bağlı kompleks korpus
kallosum agenezi.
Genetik Görülme Sıklığı
Genel olarak L1 sendromunun prevalansı
bilinmemektedir; Ancak, HSAS’ın 30.000 erkekten 1’ini etkilediği tahmin
edilmektedir.
Kalıtım Paterni/ Deseni
L1 sendromuna, esas olarak gelişen
sinir sisteminde ifade edilen L1 hücre adezyon molekülünü kodlayan L1CAM
genindeki (Xq28) mutasyonlar neden olur. Geniş klinik spektrumun açıklanmasıyla
bugüne kadar 240’dan fazla farklı mutasyon bildirilmiştir. Mutasyonların
yaklaşık% 7’sinin de novo olduğu rapor edilmiştir.
Bu durum X’e bağlı bir desende
kalıtsaldır. Bozukluğa neden olan mutasyona uğramış gen, her bir hücrede
bulunan iki cinsiyet kromozomundan biri olan X kromozomunda bulunuyorsa, X ile
bağlantılı bir durum olarak kabul edilir. Sadece bir X kromozomu olan
erkeklerde, her hücrede genin tek kopyasındaki bir mutasyon, duruma neden olmak
için yeterlidir. X kromozomunun iki kopyasına sahip olan kadınlarda, her
hücrede genin değiştirilmiş bir kopyası durumun daha az ciddi özelliklerine yol
açabilir veya hiçbir belirti veya semptomlara neden olmayabilir. X’e bağlı
kalıtımın bir özelliği, babaların X’e bağlı özellikleri oğullarına
geçirememeleridir.
Teşhis Yöntemleri ve Tedavileri
Tanı yöntemleri: Erkek hastalarda tanı, karakteristik klinik ve nöropatolojik bulgular ve X-bağlantılı bulaşma ile tutarlı bir aile öyküsü temelinde yapılır. Manyetik rezonans görüntüleme (MRG) veya otopsi ile tespit edilen piramitlerin iki taraflı yokluğu, pratikte, sendromun patognomonik bir özelliğidir. Teşhis L1CAM geninin moleküler genetik testi ile doğrulanabilir.
Ayırıcı tanı: Diğer hidrosefali ve spastik parapleji bozuklukları ekarte edilmelidir. Pediatrik / nörolojik / klinik genetik çalışmaları, olası bireysel hastalıkların teşhisini sağlar.
Doğum öncesi tanı: Bir aile üyesinde L1CAM hastalığına neden olan bir mutasyon tespit edilmişse, dişi taşıyıcılarda prenatal test yapılabilir. Koryon villus örneklemesi veya amniyosentez ile elde edilen hücrelerde kromozom analizi ile fetal cinsiyetin belirlenmesinden sonra, fetal hücreler bilinen hastalığa neden olan mutasyon için taranabilir. Kızlar etkilenebilir ve kadın fetüslerde 20 haftada ultrason önerilir. Bununla birlikte, 20 haftadaki normal fetal ultrason bozukluğu ekarte etmez: bu aşamada hidrosefali yokluğu erkek fetüsün etkilenmediğini garanti etmez.
Belirtilerin tedavisi:
L1 sendromunun tedavisi, her bir
bireyde belirgin olan spesifik semptomlara yöneliktir. Pediatri, çocuk
nörolojisi, beyin cerrahisi, rehabilitasyon ve klinik genetik alanlarında
uzmanlığa sahip çok disiplinli bir ekibi dahil etmek en iyi yöntemdir. Beyin
omurilik sıvısının (BOS) şantlanması, kafa içi basıncı azaltmak için gerektiği
gibi yapılmalıdır. Bazı hafif vakalarda, tendon transferi ve / veya atel
başparmak fonksiyonunu iyileştirebilir. Uyarılmış başparmaklar için düzeltici
cerrahi belirtilmemiştir. Zihinsel engellilik oldukça değişkendir ve gelişimsel
ilerleme izlenmeli ve danışmanlık verilmelidir. Erken müdahale L1 sendromlu
çocukların potansiyellerine ulaşmalarını sağlamak için önemlidir. Etkilenen
çocuklara faydalı olabilecek özel hizmetler arasında özel eğitim, özel sosyal
destek, fizik tedavi ve / veya diğer tıbbi, sosyal ve / veya mesleki hizmetler
yer alabilir. Genetik danışmanlık, etkilenen bireylerin aile bireyleri için
önerilmektedir.
Prognoz: Hidrosefali erken bebeklik döneminde ölü doğum veya ölüme neden olabilir. Prognoz, tezahürlerin ciddiyetine bağlıdır.
Hastalığın Diğer İsimleri
eklenmiş başparmak-zihinsel gerilik sendromu
uyarılmış başparmak-zeka geriliği sendromu
corpus callosum hypoplasia, mental retardation, adducted thumbs,
korpus kallosum hipoplazisi, zihinsel gerilik, bağımlı baş parmaklar,
Spinocerebellar
ataksi tip 2 (SCA2), hareket ile ilgili aşamalı olarak ilerleyen problemlerle
karakterize bir durumdur. Bu durumu olan insanlar başlangıçta koordinasyon ve
denge (ataksi) ile ilgili sorunlar yaşarlar. SCA2’nin diğer erken belirti ve
semptomları, konuşma ve yutma güçlüğü, sertlik, titreme ve göz hareketini
kontrol eden kaslardaki zayıflığı (oftalmopleji) içerir. Göz kası zayıflığı
hızlı göz hareketleri yapma yeteneğinin azalmasına neden olur (sakkadik
yavaşlama).
Zamanla,
SCA2’li bireyler uzuvlarda (periferik nöropati), kas kaybı (atrofi), kontrolsüz
kas gerilmesinde (distoni) ve istem dışı sarsıntı hareketlerinde (kor) duyu ve
zayıflık gelişebilir. SCA2’li bireyler kısa süreli hafıza, planlama ve problem
çözme ile ilgili problemlere sahip olabilir veya entelektüel işlevde (demans)
genel bir düşüş yaşayabilirler.
Hastalığın
belirti ve semptomları tipik olarak yetişkinlik çağında başlar, ancak
çocukluktan geç yetişkinliğe kadar herhangi bir zamanda ortaya çıkabilir.
SCA2’li insanlar genellikle semptomların ortaya çıkmasından sonraki 10 ila 20
yıl hayatta kalırlar.
Hastalığın Nedeni
ATXN2
genindeki mutasyonlar SCA2’ye neden olur. ATXN2 geni, ataksin-2 adlı bir
proteini yapmak için talimatlar sağlar. Bu protein vücutta bulunur, ancak
işlevi bilinmemektedir. Ataksin-2, hücrelerin içindeki sıvıda (sitoplazma)
bulunur, burada endoplazmik retikulum adı verilen bir hücre yapısıyla
etkileşime girdiği görülür. Endoplazmik retikulum, protein üretimi, işlenmesi
ve taşınmasında rol oynar. Araştırmacılar ataxin-2’nin, DNA’nın kimyasal bir
kuzeni olan RNA’nın işlenmesinde rol oynayabileceğine inanıyor. Ataksin-2’nin,
RNA’dan proteinlerin üretiminde de rol oynadığı düşünülmektedir (DNA’nın
genetik bilgisinin çevirisi).
SCA2’ye
neden olan ATXN2 gen mutasyonları, bir CAG trinükleotit tekrarı olarak bilinen
bir DNA segmentini içerir. Bu segment, arka arkaya birçok kez görünen bir dizi
üç DNA yapı bloğundan (sitozin, adenin ve guanin) oluşur. Normal olarak, CAG
segmenti gen içinde yaklaşık 22 kez tekrarlanır, ancak herhangi bir sağlık
sorununa neden olmadan 31’e kadar tekrar edilebilir. ATXN2 geninde 32 veya daha
fazla CAG tekrarı olan kişiler SCA2 geliştirir. 32 veya 33 tekrarı olan kişiler
ilk yetişkinlik döneminde SCA2’nin belirti ve semptomlarını yaşamaya
meyilliyken, 45’den fazla tekrarı olan kişilerin genellikle gençleri tarafından
belirti ve semptomları vardır.
Anormal
derecede uzun olan CAG segmentinin ataksin-2 proteinin fonksiyonunu nasıl
etkilediği açık değildir. Anormal protein görünüşte hücre ölümüne yol açar,
çünkü SCA2’li insanlar beynin farklı bölgelerinde beyin hücrelerinin kaybını
gösterirler. Zamanla, beyin hücrelerinin kaybı, SCA2’nin karakteristik hareket
problemlerine neden olur.
Genetik Nedenler
Bu
durum otozomal dominant paternde kalıtsaldır, bu da her hücrede değiştirilmiş
genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir.
Etkilenen bir kişi genellikle değiştirilmiş geni, etkilenen bir ebeveynden
alır. Ancak, SCA2’li bazı kişilerin, bozukluğu olan bir ebeveyni yoktur. ATXN2
geninde CAG tekrarında artış olan ancak tekrarlayan SCA2 olmayan bireyler,
bozukluğu geliştirecek çocuk sahibi olma riski altındadır.
Değiştirilmiş ATXN2 geni, bir nesilden diğerine geçerken, CAG trinükleotit tekrarının uzunluğu sıklıkla artar. Daha fazla sayıda tekrar, genellikle daha erken belirti ve semptomların başlangıcı ile ilişkilidir. Bu olguya beklenti denir. Beklenti, ATXN2 geni, bir kişinin babasından (baba mirası), bir kişinin annesinden miras aldığından (maternal miras) miras kaldığında daha belirgin olma eğilimindedir.
Görülme Sıklığı
SCA2
prevalansı(yaygınlığı) bilinmemektedir. Bu durumun en sık görülen
spinocerebellar ataksi tiplerinden biri olduğu tahmin edilmektedir; bununla
birlikte, tüm spinocerebellar ataksi tipleri nispeten nadirdir. SCA2, Küba’da,
özellikle 100.000 kişiden yaklaşık 40’ının etkilendiği Holguín eyaletinde daha
yaygındır.
*Ayak, kulak, parmak gibi vücudun çevresel bölümlerini
etkileyen herhangi bir etki, hastalık, patolojik değişim.
Diğer İsimleri
APSS
Akral tip
Özet
Acral peeling
cilt sendromu, cildin üst tabakasının ağrısız soyulmayla karakterize bir cilt
hastalığıdır. “Acral” terimi, bu durumda cildin soyulması, eller ve
ayaklarda en belirgin olduğu anlamına gelir. Bazen, kol ve bacaklarda da
soyulma meydana gelir. Soyulma genellikle doğumdan belirgindir, ancak durum
çocuklukta veya daha sonra yaşamda da başlayabilir. Cilt soyulması ısıya, neme
ve diğer nem formlarına ve sürtünmeye maruz kalmasıyla daha da kötüleşir. Altta
yatan cilt geçici olarak kırmızı ve kaşıntılı olabilir, ancak genellikle iz
bırakmadan iyileşir. Acral peeling cilt sendromu, diğer sağlık problemleriyle
ilişkili değildir.
Hastalığın Tespiti
İyi kayıtlı
veriler ve fizik muayene sıklıkla tanı koymak için yeterli olsa da, etkilenen
dokunun cerrahi olarak çıkarılması ve mikroskopik değerlendirmesi (biyopsi) gibi
özel testler zaman zaman gerekli olabilir. Büyük cilt tabakalarının sürekli
dökülmesi, soyma cilt sendromunu Netherton sendromundan ve konjenital
iktiyoziform eritroderma gibi diğer otozomal resesif konjenital iktiyoz
tiplerinden ayırt eder. “Kolodion bebekler” denilen cilt, birkaç hafta sonra
soyulur ve belirtileri zaman geçtikçe tekrar ortaya çıkan soyma cilt sendromu
hastalarının aksine geri dönmez.
Görülme Sıklığı
Akral peeling
cilt sendromu, tıp literatüründe bildirilmiş birkaç düzine vaka ile nadir bir
durumdur. Bununla birlikte, belirtileri ve semptomları hafif ve diğer cilt
hastalıklarınınkilere benzer olma eğiliminde olduğundan, durum muhtemelen tanı
almaz.
Hastalığın Nedeni
Akral peeling
cilt sendromuna TGM5 genindeki mutasyonlar neden olur. Bu gen, cildin dış
katmanının (epidermis) bir bileşeni olan transglütaminaz 5 adı verilen bir
enzimin hazırlanmasına yönelik talimatlar sağlar. Transglutaminaz 5, epidermal
hücreleri çevreleyen ve cildin vücut ile çevresi arasında koruyucu bir bariyer oluşturmasına
yardımcı olan kornifiye hücre zarfı adı verilen bir yapının oluşumunda kritik
bir rol oynar.
TGM5 gen
mutasyonları, transglutaminaz 5’in üretimini azaltır veya hücrelerin bu
proteinden herhangi birini yapmalarını önler. Bir transglütaminaz 5 kıtlığı,
epidermisin en dıştaki hücrelerinin altta bulunan deriden kolayca ayrılmasını
ve soyulmasını sağlayan kornifiye hücre zarfını zayıflatır. Bu soyma, ellerde
ve ayaklarda en belirgindir, çünkü muhtemelen bu alanlar neme ve sürtünmeye
aşırı derecede maruz kalma eğilimindedir.
Tedavi
Cildi yumuşatıcı
merhemler uygulayarak cilt sendromunu tedavi etmek, özellikle cilt nemliyken
bir banyodan sonra biraz rahatlama sağlayabilir. Sade petrol jölesi veya
vazelin tercih edilir. Kortikosteroidlerin ya da sistemik retinoidlerin (A
vitamini türevleri) hiçbiri gösterilmemiştir ya da etkili değildir ve hepsinin
ciddi yan etkileri ya da ters reaksiyonları olabilir.
Genetik Etkenler
Bu durum otozomal
resesif bir kalıtsal kalıtımla ifade edilir, yani her bir hücredeki genin her
iki kopyası da mutasyonlara sahiptir. Otozomal resesif hastalığı olan bir
bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır,
ancak bunlar genellikle durumun belirtilerini ve semptomlarını göstermezler.
Spinoserebellar Ataksi Tip 3-Machado
Joseph Hastalığı
Spinoserebellar ataksi tip 3(SCA 3) harekette
ilerleyici problemlerle karakterize bir durumdur.Bu duruma sahip insanlar
başlangıçta(ilk başlarda) koordinasyon ve dengeyle ilgili(ataksi) problemler
deneyimlerler.Diğer erken işaret ve semptomları konuşma zorlukları,kontrol
edilemeyen kas gerginlikleri(distoni),kas katılığı(spastisite),rijidite,tremorlar,şişkin(pörtlek)
gözler ve çift görüşü içerirler.Bu duruma sahip insanlar huzursuz bacak
sendromu yada REM uykusu hastalığı gibi uyku hastalıklarını
deneyimleyebilirler.Huzursuz bacak sendromu bacaklarda hissizlik yada
karıncalanmayla birlikte hissi durdurmak için bacakları hareket ettirme
dürtüsüyle karakterize bir durumdur.REM uyku davranışı hastalığı uykunun rüya
görme aşamasında(REM) aktif olan kaslarda olan bir durumdur,etkilenmiş insanlar
sıklıkla rüyalarını canlandırırlar.Bu uyku hastalıkları bireylerde gün boyunca
yorgun hissetme etkisi bırakma eğilimindedirler.
Zaman geçtikçe SCA3’lü bireylerde uzuvlarında
güçsüzlük ve duyu kaybı(periferal nöropati),kas krampları,kas
seğirmeleri(fasikülasyonlar) ve yutma zorlukları gelişebilir.SCA 3’lü bireyler hafıza,planlama ve problem çözmede
problem yaşayabilirler.
Bu hastalığın işaret ve semptomları tipik
olarak orta-yetişkinlikte başlar ama çocukluktan geç yetişkinliğe kadar
herhangi bir zamanda da ortaya çıkabilir.SCA3’lü insanlar er yada geç
tekerlekli sandalyeye gerek duyarlar.Genellikle semptomlar ilk ortaya çıktıktan
sonra 10 veya 20 yıl hayatta kalabilirler.
Sebepleri
ATXN3 genindeki mutasyonlar SCA3’e sebep
olur.ATXN3 ataksin-3 denen enzimin yapımı için,ki bu vücuttaki tüm hücrelerde
bulunur,talimatları sağlar.Ataksin-3 ubikuitin-proteazom sistemi denen aşırı
yada zarar görmeyi yada onlardan kurtulmayı sağlayan mekanizmayı
içerir.Ubikuitin molekülü ihtiyaç olmayan proteinlere
ilişmiştir(bağlı),hücrelerin içinde bozulacak(değeri düşürülmüş) proteinleri onlara etkiletler.Ataksin-3 bu
istenmeyen proteinlerin değeri düşmeden önce ubikuitini uzaklaştırır ki
ubikuitin tekrar kullanılabilsin.Araştırmacılar ayrıca ataksin-3’ün protein
üretiminin(transkripsiyon) ilk aşamasını düzenlemeyi de içerdiğine inanıyorlar.
ATXN3 gen mutasyonları CGA trinükleoit
tekrarı içerdiği bilinen DNA segmentinde SCA 3’e sebep olurlar.Bu segment 3
seri DNA yapım bloklarının(dsitozin,adenin ve guanin) düzenlemesi dizide çok
kez ortaya çıkar.Normalde,CAG segmenti genin içinde 12 ila 43 kez tekrar
edilir.Çoğu insan 31 CAG tekrarından azına sahiptirler.44 ila 52 CAG tekrarlı
insanlar “orta seviye tekrara sahip” olarak tanımlanmıştır. Bu
bireylerde SCA3 gelişebilir ya da gelişmeyebilir.75 yada daha az tekrara sahip
insanlarda orta yetişkinlik döneminde SCA3 ‘ün işaret ve semptomlarının ilk
deneyimlerine sahip olabilirken 80 civarında tekrara sahip olan insanlarda
gençliklerinde işaret ve semptomlarına sahip olma eğilimindedirler.
CAG segmentinin uzunluğunun artması ataksin-3
enziminin anormal uzun versiyonunun üretiminin
kıvrılarak yanlış 3-boyutlu şekle dönüştürmeye yönlendirebilir.Bu
fonksiyonel olmayan ataksin-3 artık ihtiyaç olmayan proteinlerden ubikuitini uzaklaştıramaz.Sonuç
olarak,bu istenmeyen proteinler,ubikuitin ve ataksin-3 ile birlikte,hücre
çekirdeğinin içerisinde dizileri kümeler(kümeleşme).Bu kümeleşmelerin hücre
fonksiyonunu nasıl etkilediği belirsizdir çünkü sağlıklı hücrelerde içinde
bulunur bir taraftan da bunlar ölürler.
Sinir hücreleri(nöronlar) ve diğer tip beyin
hücreleri ATXN3 genindeki mutasyonlardan en çok etkilenenlerdir.SCA3 beynin
beynin spinal korda(beyin sapı) bağlı bölümünde,beynin hareketlerin
koordinasyonunu içeren bölümünde(serebellum) ve beynin diğer alanlarıyla hücre
ölümüyle bağlantılıdır.Bu durumun ayrıca spinal korddaki nöronların ölümüyle
ilişkilidir.Zamanla,spinal kordda ve beyinde hücre kaybı SCA3’ün karakteristik
işaret ve semptomlarına sebep olur.
İşaret ve Semptomlar
MJD Tip 1 semptomları 10 ve 20 yaşları arasında
ortaya çıkar ve hızla ilerler.Kollarda ve bacaklarda şiddetli
güçsüzlük(distoni),kas rijiditesi veya spastisitesi(hipertoni),beceriksiz vücut
hareketleri(ataksi) sıklıkla yavaş,sersemletici,sallanarak yürüyüş(atetoz)
yanlışlıkla sarhoşluk için,geveleyerek konuşma ve yutkunma(dizartri) ve göz
hareketlerini kontrol eden kaslara olası zarar verme(oftalmopleji) ve pörtlek
gözler semptomlarını içerirler.Ussal uyanıklık ve entelektüel kapasite
etkilenmemiştir.
MJD Tip 2 semptomları Tip 1’le benzerdir ama
tip 1’e oranla hastalık genellikle daha yavaş
ilerler.Tip 2 hastalığının başlangıcı 20 ve 50 yaş arasındadır.Tip 2’nin
ayırıcı karakteristiği ise artmış serebellum disfonksiyonu sonucu düzensiz
yürüyüş(ataksi) ve kollar ve bacakların hareketinin birlikte koordinasyonunda
spastik kas hareketlerinde zorluktur.
MJD Tip 3 yaşamda 40 ve 70 yaş arasında
ortaya çıkar ve düzensiz yürüyüş(ataksi) ve bu hastalığın diğer formlarından
periferal sinirlerin inflamasyonu ve dejenerasyonu(motor polinöropati) yüzünden
kas kitlesi kaybıyla(amyotrofi) ayırt edilebilmesiyle karakterizedir.His
kaybı,acıya hassasiyetin yokluğu,anormal duyular,kolların ve bacakların
koordineli hareket yeteneğinde bozulma ve diyabet ayrıca yaygındır.Tip 3
hastalığının ilerleyişi 3 tipin en yavaşıdır.
Sıklık
SCA3’nın prevelansı bilinmiyor.Bu durumun
spinoserebellar ataksinin en yaygın tipi olduğu düşünülüyor yinede tüm
serebellar ataksi tiplerine oranla nadirdir.SCA3 Aborjin popülasyonu gibi
belirli popülasyonlarda yaygın olarak ortaya çıkar.
Machado-Joseph Hastalığının Diğer
İsimleri
Otozomal Dominant Serebellar
Dejenerasyon
Azorean Nörolojik Hastalık
Azorean Ataksisi
Joseph Hastalığı
Machado Hastalığı
MJD
Spinoserebellar Ataksi Tip 3(SCA3)
Striatonigral Dejenerasyon,Otozomal
Dominant Tip
Nigrospinodentatal Dejenerasyon
Machado-Joseph Hastalığının
Altdalları
Machado-Joseph Hastalığı Tip 1(MJD 1)
Machado-Joseph Hastalığı Tip 2(MJD 2)
Machado-Joseph
Hastalığı Tip 3(MJD 3)
Kalıtım Paterni
Bu durumun
kalıtımı otozomal dominant paterndir,bu her hücrede değiştirilmiş genin bir
kopyasının hastalığa sebep olmasına uygundur demektir.Çoğu olguda etkilenmiş
kişinin ebeveynlerinden birisi bu duruma sahiptir.
Değiştirilmiş
ATXN3 geni gelecek jenerasyona öncekinden aktarılır,CAG trinükleotitinin tekrarının
uzunluğu sıklıkla artar.Çok sayıda tekrarlar işaret ve semptomların genellikle
erken başlangıcı ve daha hızlı ilerleyişle bağlantılıdır.Bu fenomene beklenti
denir.Beklenti ATXN3 geninin kişinin babası tarafından aktarılmasının(baba
tarafından kalıtım) kişinin annesi tarafından aktarılmasından(anne tarafından
kalıtım) daha belirgin olmasıdır.
Nadir
olgularda,ATXN3 geninin genişletilmiş CAG tekrarlarının bireylerde her
hücredeki iki kopyada da bulunduğu rapor edilmiştir.Bu insanlar sadece tek
mutasyonlu kişilerden daha şiddetli işaretler ve semptomlar gösterme
eğilimindedirler ve bu durumun özellikleri çocuklukta ortaya çıkar.
Standart Terapiler
Teşhis
Aile
hikayesi ve fiziksel muayene yardım ederken olguların %100’ünde altın standart
hastanın DNA’sında şüpheli CAG üçlülerinin direkt tanınıp tespit edilmesidir.Bu
özelleşmiş genetik klinik laboratuvarlarında hazır olarak yapılabilir.
Tedavi
Tedavi
semptomatik ve destekleyicidir.L-dopa ve baklofen hapları kas sertliğini ve
spastisitesini azaltabilir.Bu hastalığın teşhisi konan bireylerin aile
üyelerinden en az birisi gen danışmanlığını dikkate almalıdır.
Genel Bilgi Balo Hastalığı Multipl Skleroz’un (MS) nadir görülen ve ilerleyen bir çeşididir. Genellikle yetişkinlikte görülür, ancak çocukluk vakaları da bildirilmiştir. Multipl skleroz tipik olarak bal mumlu ve zayıflayan bir hastalık olmasına rağmen, Balo Hastalığı hızlı ilerici olma eğiliminde olduğundan farklıdır. Belirtileri baş ağrısı, nöbetler, kademeli felç, istemsiz kas spazmları ve bilişsel kaybı içerebilir. Balo hastalığı, beyindeki sinir liflerinin etrafındaki yağ örtüsünün kaybı ve adrenal bezin ilerleyici dejenerasyonu ile karakterize nadir görülen kalıtsal bir metabolik hastalıktır.
Belirti ve Semptomlar Semptomlar, beynin etkilenen bölgelerine göre değişir. Belirtiler birkaç hafta içinde hızlı bir şekilde veya iki ila üç yıl boyunca daha yavaş ilerleyebilir. Bu bozukluğun semptomları arasında genelleştirilmiş kas güçsüzlüğü (hipotoni), hareketi koordine etme yeteneğinin (ataksi), spastik parsiyel felç ve/veya kollarda veya bacaklarda karıncalanma veya yanma hisleri abartılı refleks yanıtları (hiperrefleksi) olabilir. Çoğu vaka, kas spazmları ve felç dahil olmak üzere, daha sık görülen MS tipinde bulunabilecek semptomların kademeli başlangıcı ile karakterizedir. Etkilenen ve zihinsel bozulma yada fizyolojik anormallikleri içerebilen beyin bölgelerine bağlı olarak diğer nörolojik semptomlar gelişir. Bununla birlikte, en ciddi haliyle Balo Hastalığı, yüksek ateş ve ağrılı baş ağrılarıyla başlayan bulaşıcı bir hastalığın varlığını da önerebilir.
Genetşk Görülme Sıklığı Balo’nun hastalığı Asya’da, özellikle Çin ve Filipinler’de yaşayan insanlar arasında en yaygın olanıdır. Yetişkinlerin çocuklardan daha fazla kazanması olasıdır ve hem erkekleri hem de kadınları etkileyebilir. İnsanlar genellikle 30’lu yaşlarda hastalığı yakalarlar. Kalıtım Deseni Doktorlar, Balo hastalığını neyin tetiklediğinden emin değil. Vücudunuzun yanlışlıkla sağlıklı dokulara saldırıp şişmesine veya iltihaplanmasına neden olan bir tür otoimmün durum olduğunu düşünüyorlar. Balo hastalığına yakalanan bazı kişiler, diğer semptomları fark etmeden hemen önce yüksek ateşli ve şiddetli baş ağrıları olan bir hastalığa sahiptir. Bu nedenle, doktorlar kesin olarak bilinmese de, bunun bir enfeksiyonla bağlantılı olabileceğini düşünüyorlar.
Teşhis Yöntemleri ve Tedavileri Balo hastalığı çok nadir olduğu için, beyin ve sinir sistemi (nörolog) problemlerinde uzmanlaşmış bir doktora görünmek en iyisidir. Tıbbi geçmişini ve semptomlarını incelemek gerekiyor. Ayrıca ne kadar iyi hareket ettiğinizi ve kasların bazılarının diğerlerinden daha zayıf olup olmadığını görmek için fiziksel bir muayene edilecektir ve hafızanızın ne kadar iyi olduğunu ne kadar iyi konuşulduğunu kontrol edilecek. Muhtemelen, lezyonları kontrol etmek için beyninizin ve omuriliğinizin manyetik bir rezonans görüntüleme (MRG) taramasını da önerilecek. Vücudunuzun iç kısmının ayrıntılı resimlerini yapmak için güçlü mıknatıslar ve radyo dalgaları kullanır. Ayrıca bir enfeksiyon olup olmadığını kontrol etmek için kan testleriniz olabilir veya doktorun test için belden küçük miktarda spinal sıvı alınabilir. Bazı durumlarda, doktorun uyarılmış bir potansiyel (EP) testi önerebilir. Hastalığın Diğer İsimleri Balo Hastalığı Concentric Sclerosis Encephalitis Periaxialis Concentrica Leukoencephalitis Periaxialis Concentric Adrenolökodistrofi Kaynakça https://rarediseases.org/rare-diseases/balo-disease/ https://www.webmd.com/multiple-sclerosis/balos-disease https://pdfs.semanticscholar.org/ce23/63690425e3e9d502237cb393bddb54d1cf60.pdf
Baller-Gerold
sendromu (BGS) doğumla beraber görülen nadir bir hastalıktır.
Bu
hastalığın otozomal resesif paterne sahip olduğu düşünülmektedir. Belirli kafa
tası kemiklerinin erken kaynaşması (kraniosinostosiz) ile radyal ray
anormallikleri olarakta bilinen el ve dirsek kemiklerinde de anormal karakter
göstermektedir.( “Radyal ray anormalliği” medikal dilde el ve dirseklerde
görülen büyüme bozuklukları ifade etmektedir.)
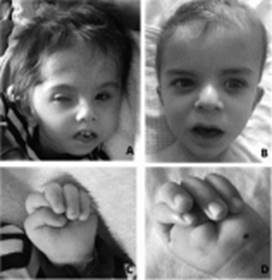
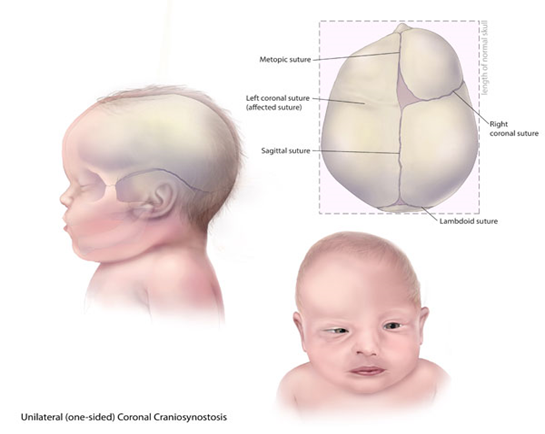
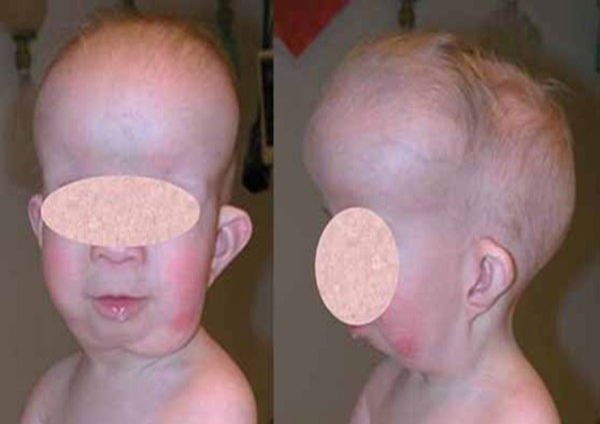
Baller-Gerold Sendromuna yaşayan insanlar erken genellikle, kafatasının
kenarına ait kafa kemiklerinin dikişe benzeyen ek yeri [1] ile bir kulaktan
başlayıp diğerine belirli bir büyüme çizgisi ile devam eden kaynaşmış kafa tası
kemiklerine sahiplerdir.
Bu değişimler abnormal şekillenmiş bir baş yapısı, öne çıkıntılı alın [2], dar
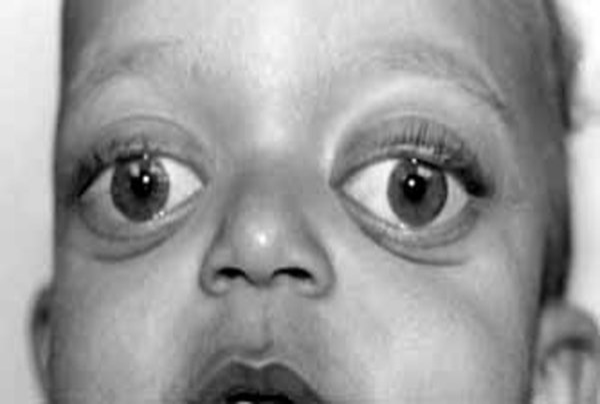
göz yuvasına sahip öne doğru gelen gözlere (ökülar proptozis) [3] neden
olmaktadır.Diğer belirgin yüz hatları ise arası açık (birbirinden uzak) gözler
(hipertelorizm) [4], küçük bir ağız ve eyer biçimli ya da az gelişmiş bir
burundur. Baller-Gerold sendromuna sahip bireyler boyda kısalık, hatalı oluşmuş
ya da eksik diz kapağı (patellae) gibi çocukluk
döneminde başlayan büyüme bozukları ve daha bir çok belirti ve semptomlara
sahiptir. Ayrıca doğumdan bir kaç ay sonra kollar ve bacaklardaki deride
kızarıklık
İle döküntü görülüp bu zaman geçtikçe vücuda dağılmaktadır.Bu dağılım sebebi ile deride bölgesel renk değişimleri, bazı bölgelerde deri incelmesi ve kan damarlarının deri altında küçük çaplı kümelenmelerine yol açar.( Bu deri problemleri toplu olarak poykilodermi olarak adlandırılırlar.)
Figur [1]
Figür [2]
Figür [3]
Figür [4]
Baller-Gerold sendromu RECQL4 adlı DNA stabilizasyonunu sağlayan genin mutasyonuyla oluşur. Aynı zamanda doğumdan önce sodyum valproate adlı bir ilaç kullanımıyla da ilişkilendirlirmektedir. Hamilelik sırasında bu ilacın kullanımı sonucu doğan bebeklerde Baller- Gerold sendromuna benzer anormallikler tespit edilmiştir. Fakat ilacın Baller- Gerold hastalığına yol açtığı kesinleştirilmemiştir.
Belirti ve Semptomlar
Baller-Gerold Syndromu ile doğan çocuklarda eklem ve kafatadı kemiklerinde
erken kaynaşma görülür. Bu başın yukarı yönde büyümesine ve başın koni şekli
gibi sivri görülmesine sebep olur.
Vücutta ise uzun ön kol kemiğinin serçe parmağa uzanan kısmında kısalık ve
eğrilik, baş parmak kısmında ise az gelişmişlik ya da eksiklik görülebilir. Bu
iskeletsel anormallikler vücudun iki tarafında da seyredebilir (Simetrik).
Bazen eldeki karpal ve metakarpal olarak adlandırılan kemiklerde eksiklik ya da
farklı oluşma gözlemlenbilir. Dik kapağının az gelişmişliği ya da eksikliği de
görülebilir. Bazı BGS vakalarında omurilik ve leğen kemiğinde anormallikler
görülebilir. BGS vakalarında büyüme bozukluğu görülmektedir ve çocukluk
döneminden başlayan bu bozukluk boy kısalığı ve kilonun normalin altında
seyretmesine yol açabilir. Aynı zamanda BGS vakalarında kalp bozuklukları da
görülebilmektedir. Bazen bu kalp
bozuklukları ameliyat gerektirse de, bazı vakalar zaman ile amelyat
gerekmeksizin geçmektedir.
Baller-Gelord sendromunda zeka geriliği vakaları görülse bile çoğu birey
normal zekaya sahiptir.
Bazı BGS vakalarında anüs olduğundan daha önde olabilir (ön tarafa
yönelmiş) ve bazen anüs eksikliği ya da kapalı olma durumu olabilir. Bu durum
genital bölge ile anüs arasında fistule bulunan anormal bağlanmaya sebep
olabilir. Bu durumlarda ameliyat ile düzeltmeler yapılabilmektedir.
BG sendromlu kişiler ya da taşıyıcıları çeşitli kanser hastalıklarına
yakalanmada yüksek riske sahiplerdir. (Deri kanseri, kemik ve lenfoma vb.)
RECQL4 geni kansere yakalanma riskini arttıran etkendir.
Genetik Görülme Sıklığı
Baller-Gerold sendromunun
yaygınlığı bilinmemekle beraber bu nafir vakanın miliyonda 1 kişiyi etkilediği
düşünülmektedir. Medikal alanda şu ana kadar 40’tan daha az vaka ile
karşılaşılmıştır.
Kalıtım Paterni/Deseni
Baller-Gerold sendromu otozomal resesif karaktere sahip kalıtsal bir hastalıktır,
bu da hastalığın oluşabilmesi için mutasyonlu iki genin bir araya gelmesi
gerektiği anlamına gelir. RECQL4 geninde oluşan mutasyon hastalığa sebep
olmaktadır.
Otozomal resesif pozisyonunda olup iki mutasyonlu geni de taşıyan vakaların
anne ve babaları mutasyonlu genden birini taşısalar bile tipik olarak
hastalığın özelliklerini göstermezler.
RECQL4 geninde oluşan mutasyon hastalığa sebep olur ve bu gen RecQ helikaz adı verilen proteinin oluşması için gereken talimatları vermektedir. (Helikazlar DNA’nın spiral şeklinde bağlanıp ayrışması için gerekli bir enzimdir.Bu DNA’daki çift zincirin ayrılması, hataların düzeltilmesi için gerekli olan DNA hazırlanmasını yapmak üzere DNA kopyalaması yapmayı sağlar ve RECQL4 proteini vücuttaki genetik bilgi ve vücuttaki hücre ve onarım gibi olayların stabilize edilmesine yardımcı olur. Bu gendeki mutasyon bahsedilen önemli vücutsal aktivitelerin yapılmasını aksatmaktadır. Baller-Gerold Sendromuna yol açab bu proteinin aktivitesinin kaybının nasıl oluştuğu ve bahsedilen çeşitli belirtilere nasıl yol açtığı bilinmemektedir.
Teşhis Yöntemleri ve Tedaviler
Baller-Gerold sendromunun teşhisi koronal
kraniosinostozis, kısıtlı gelişim, radyal ray malformasyonu (şekil bozukluğu)
ve poykilodermi gibi klinik teşhişlere bağlıdır. Kafatası kemiklerinde, önkol,
parmak eklemlerinde doğumla belirgin olan anormallikler hastalığın teşhişinde
rol oynar. Doktorlar bu anormallikleri kafa tası X-Ray’i ya da 3D-CT taraması
ile teşhiş edebilirler.
Baller-Gerold sendromu genetik RECQL4 genindaki mutasyon bulunarakta teşhis
edilebilir. Bu teşhisten sonra genetik danışmanlık ve yönetimi ile destek
alınmalıdır, çünkü genetik bir testin sonucunda hastalığın başka hangi
belirtilere yol açabileceği ve medikal riskler hakkında bilgi sahibi
olunabilir. RECQL4 genindeki mutasyon BGS’nin teşhisinde etkili olsa da yakın
zamandaki medikal kaynaklar BGS’nin belirtilerini gösteren hastaların hepsinde
bu gende görülebilir mutasyon olmadığını göstermektedir. Çoğu Baller-Gerold
Sendromunda poykilodermi ya da deride değişikliğin görülmemesi bu duruma bağlı
olabilir, farklı durumlar için farklı genetik bir sebep bulunabilir.
BGS’nin tedavisi ilk olarak kemik yapısı ve diğer
bozuklukların giderilerek kraniosinostozisten dolayı oluşan kafatası içerisindeki
basıncı ortadan kaldırmak için yapılan ameliyatlar ile sağlanır. Hastanın yaşı
ameliyatlar sırasında ne kadar küçük olursa o kadar iyi sonuçlar ortaya
çıkmaktadır.
Ayrıca BGS ye sahip bireyler için motor becerilerini
geliştirecek fizik tedavi uygulanabilir. Eğer her hangi bir kalp sorunu ortaya
çıkarsa ameliyat yapılması gereklidir.
Medikal alanda ortopedistten, dermatoloji, genetik ve beyin cerrahisine kadar
bir çok sağlık çalışanı BGS’lu bireyin tedavisinde etkin rol oynayabilir.
Diğer tedaviler ise semptomatik ve destekleyicidir.
Hastalıkla İlişkili Genler
RECQL4 genindeki mutasyon Baller-Gerold Sendromuna sebep olmaktadır. RECQL4 geni DNA stabilitesi sağlanmasında büyük rol oynar.
Akrodermatit enteropatika (AE), üç formdan
birinde meydana gelen bir çinko metabolizması bozukluğudur: doğuştan gelen
(doğuştan) bir form ve iki kazanılmış form. Doğuştan AE formu, bağırsaktan
çinko emememeye neden olan bağırsak anormallikleri ile karakterize nadir bir
genetik hastalıktır. Çinko eksikliği karakteristik olarak şu şekildedir: (1)
ağız ve / veya anüs çevresinde meydana gelen sivilce (püstüler dermatit) ile
cilt iltihabı, (2) diyare ve (3) anormal tırnaklar (tırnak distrofisi). Akut
dönemde beyin korteksinin israfı (atrofi) nedeniyle sinirlilik ve duygusal
bozukluklar belirgindir. Bu bozukluğu tanımak ve tedavi etmek önemlidir. Bu
hastalığın edinilmiş şekli benzer semptomlar oluşturur. Geçici bir form,
annenin anne sütüne çinko salgılamamasından kaynaklanabilir. Diğer kazanılmış
AE formları bazen ameliyattan sonra üst bağırsakların bir kısmını atlamaya veya
uygun miktarda çinko olmadan hazırlanan özel intravenöz beslenme programlarına
neden olur. Ek çinko genellikle belirtileri ortadan kaldırır.
Hastalığın Semptomları ve Belirtileri
Akrodermatit enteropatika, hafif veya şiddetli
olabilen kronik ishal ve dışkıda yağlı maddelerin bulunması (steatorit) ile
karakterize edilir. Konjenital formda semptomlar yavaş yavaş başlar, sıklıkla
bir bebeğin sütten kesilmesi sırasında. Ağız, anüs ve gözler gibi vücut
açıklıklarının etrafındaki deri ile dirsek, diz, el ve ayaklardaki cilt
iltihaplanır. Deri lezyonları genellikle kabarır (vezikobullous) ve kuruduktan
sonra sedef benzeri hale gelirler. Tırnakların etrafındaki deri de iltihaplı
olabilir ve tırnak yetersiz beslenmiş doku nedeniyle anormal olabilir. Saç
derisinde, göz kapaklarında ve kaşlarda saç dökülmesi toplam olabilir
(alopesi). Genellikle göz kapağını (konjonktiviti) kaplayan zarın
iltihaplanması da meydana gelir.
Bu hastalığın doğuştan formu olan
kişilerde kan çinko seviyesi, nadiren normal kan çinko seviyeleri de gözlense
de, anormal derecede düşüktür.
Bebeklerde ayrı bir geçici çinko eksikliği
türü, doğuştan gelen farklı bir anormallikten kaynaklanabilir – fakat biri
bebekte değil annede. Özellikle emziren bazı kadınlarda, pankreas tarafından
üretilen ve insan sütünde mevcut olan çinko bağlayıcı bir faktör
bulunmayabilir. Bu kadınların emzirilen bebekleri de, bu bozukluğun diğer
semptomlarıyla birlikte kanda çinko seviyelerini düşürebilir, çünkü süt, çinko
bağlayıcı faktörün uygun miktarında yetersizdir. Bebeğin diyetine alternatif
bir oral çinko kaynağı eklendiğinde (örneğin doktor tavsiyeli hazır bebek sütü) çinko eksikliği giderir ve
bebek tedavi edilir.
Tedavi ile, akrodermatit enteropatica
hastalarının tümü normal yaşam sürdürebilir
Genellikle ergenlik döneminde başlayan
uzun süreli remisyonlar ortaya çıkabilir. Bununla birlikte, nadir durumlarda,
kadınlar hamilelik sırasında hastalığın tekrarını yaşayabilir ve daha fazla
çinko takviyesi gerekebilir.
Genetik Faktörler
Akrodermatit enteropatinin konjenital
formu otozomal resesif genetik bir hastalık olarak bulaşır. SLC39A4 genindeki
mutasyonların sonucu gibi görünüyor.
Genetik hastalıklar, baba ve anneden
alınan kromozomlar üzerindeki belirli bir özellik için genlerin bir
kombinasyonu ile belirlenir.
Resesif genetik bozukluklar, bir birey her
bir ebeveynden aynı özellik için aynı anormal geni aldığında ortaya çıkar. Bir
birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için
taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin
hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her
hamilelikte% 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her
hamilelikte% 50’dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu
özellik için genetik olarak normal olma şansı% 25’tir. Risk erkeklerde ve
kadınlarda aynıdır.
Tüm bireyler birkaç anormal gen taşır.
Yakın akraba olan ebeveynlerin (akraba), ilişkisiz ebeveynlerden her ikisinin
de aynı anormal geni taşıması olasılığı daha yüksektir, bu da resesif genetik
bozukluğu olan çocukların görülme riskini artırır.
Bazı kadınlar anne sütlerinde yeterli çinko seviyeleri üretmekte başarısız olurlar – ve bu genetik bir neden de olabilir. SLC30A2 mutasyonundaki tek bir mutasyon anne sütünü çinkoyu azaltabilir. Bu eğilim iki gen anormallikine ihtiyaç duymaz, biri yeterlidir ve bu hastalığa sahip kişilerin yavrularına geçirme şansı% 50’dir.
Görülme Sıklığı
Arodermatit enteropatinin konjenital
formu, bebeklik döneminde başlayan nadir bir hastalıktır. İnsidans, 500.000
doğumda 1 civarındadır ve bu durum erkekleri ve kadınları eşit sayıda etkiler.
Bozukluğu olan kadın hastaların sağlıklı emzirilen bebekleri de etkilenebilir.
AE’nin elde edilen formu nadirdir, çünkü son yıllarda parenteral beslenme
rejimine çinko takviyeleri eklenmiştir, ancak elde edilen formlar,
gastro-intestinal malabsorpsiyon sendromunun daha sık olduğu Güneydoğu Asya ve
Sahra altı Afrika gibi bazı bölgelerde daha yaygın olmasına rağmen.
En Yaygın Tedavi Yöntemi
Acrodermatitis enteropathica, çinko sülfat
formundaki çinko takviyeleri ile tedavi edilir. Bu takviyeler, bozukluğun
teşhisi konduğu anda yapılmalı ve ömür boyu devam ettirilmelidir. İlaç
Diodoquin (iyodokinol) genellikle bir hafta içinde semptomları gideren başka
bir tedavi yöntemidir. Hastalığın intravenöz beslenmesinden kaynaklanması
durumunda, beslenme rejimine çinko takviyeleri eklemek, AE’nin tezahürlerini
önleyebilir ve / veya temizleyebilir.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
İktiyoziform eritroderma ve ekstremite defektleri ile konjenital hemidisplazi olarak da bilinen CHILD sendromu, tipik olarak cildi kaplayan büyük kırmızı ve iltihaplı lekelerle (eritroderma) , pul pul dökülme (iktiyoz) ile uzuvlarda gelişmede yetersizlik ya da uzuvların olmamasıyla karakterize genetik bir durumdur.
Belirti ve Semptomlar
Belirtiler normalde vücudun bir tarafında görülür. Beyin, kalp, akciğerler ve böbrekler gibi organların gelişimi de etkilenebilir.
Tıbbi literatürde daha hafif belirti ve semptomların bulunduğu birkaç vaka bildirilmiştir. Bu duruma, kolesterol yapımında rol oynayan bir enzimin üretimi için talimatlar veren bir gen olan NSDHL genindeki mutasyonlar neden olur. CHILD sendromu, X kromozomuna bağlı baskın bir biçimde kalıtsaldır ve neredeyse yalnızca kadınlarda bulunur.
Genetik Görülme Sıklığı
CHILD sendromu çok nadirdir ve literatürde 30’dan az olgu bildirilmiştir.
Kalıtım Paterni/Deseni
Bu durum X’e bağlı bir dominant kalıtım düzenine sahiptir. Mutasyona uğrayan gen, hastalığın iki cinsiyet kromozomundan biri olan X kromozomu üzerinde yer almasına neden olması durumunda bir durumun X’e bağlı olduğu düşünülür. Bir hücre, bu duruma neden olacak kadar yeterli düzeyde ise kalıtım gerçekleşir.
Çoğu CHILD sendromu vakası düzensiz olarak meydana gelir, bu da ailenin sadece bir üyesinin etkilendiği anlamına gelir. Nadiren, durum ailelerde olabilir ve anneden kıza geçer. Araştırmacılar, CHILD sendromunun neredeyse yalnızca kadınlarda meydana geldiğine inanıyor. CHILD sendromu olan sadece bir erkek rapor edilmiştir.
CHILD sendromu semptomlara dayanarak ve genetik testlerle teşhis edilir. CHILD sendromu için spesifik bir tedavi yoktur, ancak birkaç hastada kolesterol inhibitörü içeren topikal kremlerin cilt semptomlarını iyileştirdiği bildirilmiştir.
Çoğu durumda, X-ışınlarında da belirtilen tüm lezyonların çarpıcı lateralizasyonu güçlü bir tanısal ipucudur. Deri lezyonu histolojisi psoriaziyi andıran hiperkeratoz ve parakeratozu enflamatuar infiltratlarla gösterir. Bununla birlikte, vücut kıvrımlarını içeren papillomatoz lezyonlardan alınan biyopsiler, papiller dermiste lipid yüklü histiyositler şeklinde yüksek karakteristik verruciform ksantoma göstermektedir. X-ışınlarını tamamlamak ve tüm anomalileri tanımlamak için viscera, ekokardiyogram ve tam beyin MRG ultrasonu gerekir. Genetik testler tanıyı doğrular.
Akciğer ve kalp anomalileri potansiyel olarak ölümcüldür ve acil cerrahi müdahale gerektirebilir. Renal anomaliler ayrıca etkilenen böbreğin boşaltılmasını veya çıkarılmasını gerektirebilir. Ortopedik diş telleri veya düzeltici cerrahi gerekli olabilir. Kontralateral otolog deri greftleri başarıyla uygulanmaktadır. Deri lezyonları en iyi kolesterol ile birlikte lovastatin veya simvastatin içeren bir losyon veya merhem uygulamasıyla tedavi edilir.
Hastalıkla İlişkili Genler
NSDHL (Xq28), kolesterol biyosentezinden sorumlu bir proteini kodlar, mutasyonlar tipik olarak erkeklerde öldürücüdür. X inaktivasyonu, kadınlarda enzimden yoksun, embriyonik gelişimi bozan ve oldukça değişken bir anomali spektrumuna yol açan bir hücre mozaiği oluşturur.
Hastalığın Diğer İsimleri
Halk arasında Balık pulu veya balık derisi hastalığı olarak da bilinir
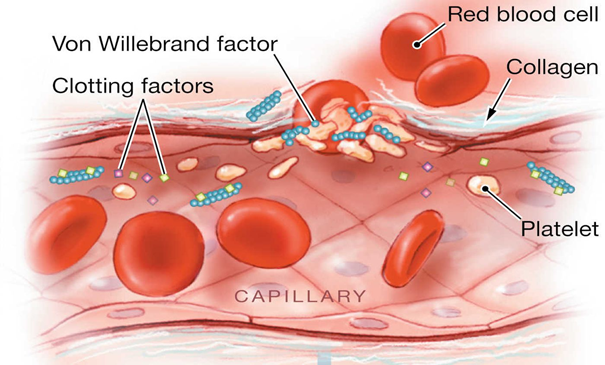
Faktör XII eksikliği, uzun bir klinik
kanama eğilimi olmadan, test tüpünde kanın uzun süre pıhtılaşmasına neden olan
nadir bir genetik kan hastalığıdır. Bir plazma proteini (glikoprotein) olan
faktör XII (Hageman faktörü) eksikliği nedeniyle ortaya çıkar. Spesifik olarak,
faktör XII, bir pıhtılaşma faktörüdür. Pıhtılaşma faktörleri, normal kanın pıhtılaşması için gerekli
olan bir grup proteindir. Bir yaralanmadan sonra, pıhtılar zarar görmüş kan
damarlarını kapatarak ve daha fazla kan kaybını önleyerek vücudu korur. Faktör
XII, genellikle hasar görmüş kan damarı duvarlarıyla temas ederek aktive
oluncaya kadar, kan dolaşımında aktif olmayan bir biçimde dolaşır. Aktivasyon
üzerine, faktör XII, koagülasyon faktörü XI ile etkileşime girer. Bu etkileşim,
bir kan pıhtısı oluşturan ek kimyasal reaksiyonlar zincirini başlatır.
Faktör XII eksikliği, herhangi bir
semptomla nadiren ilişkilidir (asemptomatik). Bununla birlikte, bir hastadan
gelen kan, pıhtılaşma süresini ölçen bir test olan parsiyel tromboplastin zaman
testine (PTT) tabi tutulduğunda, kanın pıhtılaşması çok uzun zaman alır. Bir
başka kan pıhtılaşma testi olan serum protrombin (PT) süresi de anormaldir.
Faktör XII’nin kan seviyesi büyük ölçüde değişme eğilimindedir.
Araştırmacılar, kan pıhtılaşmasına yatkın kişiler için
potansiyel bir tedavi olarak XII faktörünü bloke etmek (engellemek) için
ilaçlar üzerinde çalışıyorlar. XII’nin kan pıhtılarının gelişiminde veya
önlenmesinde ve vücuttaki genel işlevlerinde rolünün tam olarak belirlenmesi
için daha fazla araştırma gereklidir.
Tıbbi literatürde, etkilenen bazı kadınlarda faktör XII eksikliği ile tekrarlanan açıklanamayan düşükler arasında bir ilişki olduğunu öne süren raporlar da vardır.
F12 geninde, faktör XII eksikliğine
neden olan yaklaşık 20 mutasyon tanımlanmıştır. Faktör XII eksikliği, kandaki
faktör XII kıtlığı ile karakterize kalıtsal bir durumdur. Bu rahatsızlığı olan
kişiler genellikle anormal kanama veya başka semptomlar yaşamazlar. Faktör XII
eksikliği tipik olarak rutin kan testi sırasında keşfedilir, çünkü düşük faktör
XII seviyeleri kanın bir test tüpünde pıhtılaşmasının daha uzun sürmesine neden
olur. Faktör XII eksikliğine neden olan mutasyonların çoğu faktör XII’nin
yapısını değiştiren tek aminoasitleri değiştirir. Faktör XII eksikliği olan
bireylerin neden diğer pıhtılaşma faktörleri olmayanlarda olduğu gibi anormal
kanama yaşamadıkları hala açık değildir.
F12 genindeki en az iki mutasyon
kalıtsal anjiyoödem tip III ile ilişkilidir. Bu mutasyonlar, faktör XII’deki
tek protein yapı bloklarını (amino asitleri) değiştirerek proteinin
aktivitesini arttırır. Sonuç olarak, ilave sıvıların kan damarı duvarlarından
sızmasına izin veren daha fazla bradikinin üretilir. Vücut dokularında sıvı
birikimi, kalıtsal anjiyoödem tip III olan kişilerde şişme ataklarına neden
olur.
XII eksik faktörü olan (yaklaşık yarı
normal) Norveçli aileyi tanımlamıştır . Normal olmayan anormallik deneyiminin
aksine, hafif ve orta derecede kanama eğilimi ve göreceli olarak erken yaşlarda
meydana gelen yüksek serebral apopleksinin görülme sıklığı gösterdiler. Bazı
hastalarda lokal ödem, şiddetli baş ağrısı, karın ağrısı ve çeşitli alerji
şekilleri vardı. Braulke ve diğ. (1993) , azalmış faktör XII aktivitesi
seviyelerinin, tekrarlanan spontan düşükler için bir risk faktörü olabileceğini
gösteren veriler sunmuştur . Gordon ve diğ. (1981) hem pıhtılaşma aktivitesinin
hem de Hageman faktörünün antijenik özelliklerinin Oriental’lerde Amerikan
beyazlarından daha düşük olduğunu göstermiştir.
OLUŞMA NEDENLERİ (GENETİK DEĞİŞİKLER)
Faktör XII eksikliği, otozomal
resesif bir hastalık olarak kalıtsaldır. Genetik hastalıklar, biri babadan
diğeri de anneden alınan iki gen tarafından belirlenir.Resesif genetik
bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı anormal geni
aldığında ortaya çıkar.
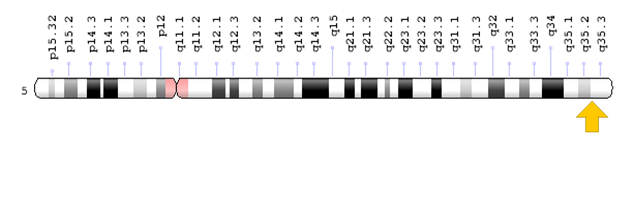
Araştırmacılar, faktör XII
eksikliğinin, kromozom 5’in (5q33-qter) uzun kolunda bulunan F12 geninin
mutasyonları nedeniyle meydana geldiğini belirlemiştir.
F12 geni, pıhtılaşma faktörü olan XII faktörü yaratır (kodlar). F12 geninin mutasyonları kanda düşük seviyelerde fonksiyonel faktör XII’ye neden olur (potansiyel olarak% 1’den az). Faktör XII’nin pıhtılaşma sürecinde oynadığı rol ve vücut üzerindeki ek etkileri tam olarak anlaşılmamıştır. Pıhtılaşma sürecine ek olarak, faktör XII’nin bir doku tamirinde ve kan damarlarının oluşumunda (anjiyogenez) rol oynadığına inanılmaktadır.
Faktör XII eksikliği, Asya kökenli
insanları diğer etnik kökenlerden daha fazla etkiler. Erkekler ve dişiler eşit
sayıda etkilenir. Hiçbir semptom genellikle faktör XII eksikliği ile ilişkili
olmadığı için, birçok kişi tanı konmamış olarak kalır. Genel popülasyondaki hastalığın
tam insidansı bilinmemektedir, ancak yaklaşık 1 milyon kişide 1 olduğu tahmin
edilmektedir.
Faktör XII eksikliği sıklıkla
ameliyattan önce yapılan rutin kan pıhtılaşması testleri sırasında yanlışlıkla
teşhis edilir. Etkilenen bireylerde, bu testler sırasında kanlarının
pıhtılaşması daha uzun sürer. Diğer testler kandaki düşük XII faktörü
seviyelerini ortaya çıkarabilir.
Klinik Test ve Çalışma
Faktör XII eksikliği tanısı klinik
belirtileri olmayan veya aktive parsiyel tromboplastin zamanı (aPTT) veya
protrombin zamanı PT) olarak bilinen tarama pıhtılaşma testleri denilen özel
testlerin anormal olduğu kanama bozukluğu öyküsü olan kişilerde şüpheli
olabilir. Bu testler kanın pıhtılaşmasının ne kadar sürdüğünü ölçer.
Bu testlerde anormal sonuçlar veren
bireyler, ancak antifosfolipid sendromu olarak bilinen bir durum için kanama
semptomları taranamaz. Edinilmiş antifosfolipid sendromu olan bireylerde
bulunan ve aPTT veya PT testlerinde benzer anormal sonuçlara neden olabilen
lupus antikoagülan adı verilen spesifik bir inhibitörü tespit etmek için bir
test yapılacaktır.
Faktör XII eksikliği teşhisi, test denilen bir test ile doğrulanabilir. Bir tahlil, pıhtılaşma faktörlerinin aktivitesini ölçen bir testtir. Faktör XII’nin eksikliğini gösterebilir.( https://rarediseases.org/rare-diseases/factor-xii-deficiency/)
Tedavi
Herhangi bir tedavi gerektirmez.
Pıhtılaşma testini düzeltmeye yönelik yapılacak plazma yerine koyma
tedavilerinin tromboz riskini artırabileceğini düşündüren veriler mevcuttur.
Hastalığın Diğer İsimleri
Pıhtılaşma
faktörü XII (Hageman faktörü) eksikliği




{kind=link}
{kind=link}
{kind=link}
{kind=link}