Genel Bilgiler,Genetik Değişiklikler/Etken
Faktörler
Johanson – Blizzard sendromu (JBS), vücudun
birçok sistemini etkileyen nadir bir hastalıktır. Hastalığın şiddeti, belirti
ve bulguları kişiden kişiye değişiklik gösterir. Çoğu semptom doğumda ya da
erken çocukluk döneminde bulgu verir. Karakteristik özellikleri pankreas
yetmezliği nedeniyle gelişen yağ ve diğer besinlerin malabsorbsiyonu, büyüme ve
gelişme geriliği, kısa boy, kalıcı dişlerde, kafatası ve yüzde anomali ve
değişen derecelerde entelektüel eksiklik ile karakterizedir.
UBR-1 genindeki mutasyona bağlı olarak gelişen otozomal resesif bir
hastalıktır. [1]
Belirti ve Semptomlar
Tabloda hastalığa sahip kişilerde
görülebilecek belirti ve semptomlar listelenmiştir. Bu semptomlar kişiden
kişiye farklılık göstermektedir. Hastalığa sahip kişilerde semptomların hepsi
birden görülmeyebilir. [2]
SEMPTOMLAR
Hastaların
%99-80’inde görülen semptomlar
Anormal saç şekliAlopesiekzokrin pankreas
yetmezliğiIntrauterin gelişme
geriliğimalabsorbsiyonkısa burunkısa boyaz gelişmiş burun
kanatları
Hastaların %79-30’unda görülen semptomlar
anormal vajina gelişimi gözyaşı noktası yokluğuanal atrezianemiönde yerleşimli anüs diş çıkmasında gecikme hipoproteinemientelektüel kapasitede azalma gözyaşı anomalileri küçük ya da az diş sensinörinal işitme kaybı
Genetik Görülme Sıklığı
Gerçek insidansı
bilinmese de görülme sıklığının 250.000 doğumda 1 olduğu düşünülmektedir.
Literatürde bildirilen 60’dan fazla vaka bulunmaktadır. Hastalık literatürde
ilk olarak Johanson ve Blizzard isimli doktorlar tarafından 1971’de
tanımlanmıştır. Etkilenen gen 2006’da Zenker ve arkadaşları tarafından
belirlenmiştir. [3]
Kalıtım Paterni
Hastalığın daha
çok kadınlarda görülmesi ve XXY kromozomlu bir erkekte bildirilen vaka
nedeniyle X’e bağlı dominant geçiş olduğu düşünülmekteydi. Otozomal resesif
kalıtım 3 aile üyesi etkilenen bir vakada Mardini ve arkadaşları tarafından
1978’de gösterildi. [4]
Teşhis Yöntemleri ve Tedaviler
Tanı
karakteristik klinik bulguların tanınması ile konur. Kesin tanı UBR-1 genindeki mutasyonun
gösterilmesiyle konur.
Prenatal tanıda hipoplazik/aplazik burun ve dilate sigmoid
kolonun görülmesi anlamlı olabilir. Moleküler test ile tanı
kesinleştirilmelidir. [5]
Semptomlara
yönelik ve destekleyici tedavi uygulanır. Pankreas yetmezliği gelişenlerde
pankreas enzim takviyesi ve besin takviyesi, vitamin eksiklikleri oluşmasını
önlemek için vitamin takviyeleri verilir. Hipotiroidi gelişen olgularda tiroid
hormon replasman tedavisi uygulanır. Baş-boyundaki ve boşaltım sistemindeki
gelişim bozuklukları ve kardiyak problemler için cerrahi tedavi
gerekebilmektedir. Sosyal ve mesleki açıdan desteklenmekte ve gerekirse özel
eğitim sağlanmaktadır. [6]
Hastalıkla İlişkili
Genler
Hastalıkla ilişkili gen 15.kromozomun
uzun kolunda bulunan UBR-1 genidir. [7]
De Barsy sendromu nadir kalıtsal bir hastalıktır ve
genellikle göz anormallikleri, büyüme geriliği, zihinsel engellilik, erken
yaşta yaşlanmış bir görüntü (prematüre yaşlanma), bol (gevşek) ve elastikliğini
kayıp etmiş bir deri (kutis laksa) ve hastadan hastaya değişiklik gösterebilen
farklı semptomlarla karakterize edilir. Ek olarak, belirgin yüz şekilleri,
iskeletsel malformasyonlar ve nörolojik (sinirsel) anormalliklerde
gözlemlenebilir. Şu ana kadar iki farklı genden en az birisinde mutasyon olması
De Barsy sendromu ile ilişkilendirilmiştir ki bu genler PYCR1 ya da ALD18A1 ancak
bazı vakalarda hasta bireyin önce bahsedilen bu iki geninde de mutasyon
olmamasına rağmen De Barsy sendromuna sahip olduklarını bilinmektedir ki bu da
bütün De Barsy sendromunu oluşmasında rol oynayan bütün genlerin
tanımlanmadığını gösterir.
Genetik Değişiklikler/
Etken Faktörler
Genel bilgi kısmında kısaca belirtiği üzere De Barsy
sendromunun bazı vakalarında PYCR1 veya
ALD18A1 genlerinden herhangi bir
tanesinde meydana gelen mutasyonlardan kaynaklandığı tespit edilmiştir. Kısaca,
nasıl bu hastalığa sebep olduğunu anlatmaya çalışıcak olursak şöyle
özetlenebilir:
Genler vücutta önemli fonksiyonlara sahip olan
proteinleri kodlamak için gerekli bilgiyi sağlarlar fakat vücudumuzdaki
herhangi bir protein üreten gende mutasyon gerçekleşirse üretilen protein
hatalı, etkisiz ve ya tamamen üretilmemiş olabilir ve o proteinin
fonksiyonlarına bağlı olarak bu hatalı üretim, birden fazla organ sistemini etkileyebilir.
Ancak bazı De Barsy sendromuna sahip hastalarda yukarıda ismi geçen bu
genlerden PYCR1 veya ALD18A1, herhangi iki genden birinde
bile mutasyona sahip olmadıkları gözlenmiştir ki bu da daha tanımlanmamış
genlerin De Barsy sendromuna etken olarak rol oynadığına işaret eder.
Belirti ve Semptomlar
Araştırmacılar çoğu zaman görülen ana semptomlar olarak
isimlendirdiği birtakım semptomlar belirleseler de semptomlar hastadan hastaya
değişiklik göstermektedir ve hala De Barsy sendromu tam olarak anlaşılmış
değildir. Bunun anlaşılmamasının sebebi birtakım nedenlere bağlanabilir ki bu
sebepler arasında şu ana kadar tanımlanmış vaka sayısını fazlasıyla az olması
ve buna bağlı olarak kliniksel olarak herhangi bir çalışmanın yapılmasının
oldukça zorlaşması, hala bilinmeyen genlerin varlığı ve etkileri olarak kısaca
özetlenebilir. Bu sebepler De Barsy sendromunun tanısının teşhis yöntemini
oldukça zorlaştırmıştır. Yukarıdaki bahsedilen sebeplerden dolayı birazdan
açıklanacak semptomlar her hastada gözlemlenmeyebilir.
De Barsy sendromuna sahip olan hastalarda oldukça geniş ve değişik semptom
grupları rapor edilmiştir. Erken yaşta yaşlanmış bir görüntü genellikle De
Barsy sendromuna sahip çocukları etkiler. Yaşlanmış görüntü çoğunlukla yüzdeki
deri ve yapıların az gelişmesinden dolayı kaynaklanır ve ek olarak bol, sarkan
elastikliğini kayıp etmiş, kutis laksanın karakterize olmuş özelliklerindendir,
deri de yüksek oranda yaşlanmış görüntüye sebep olmasında rol oynar. Az oranda
olmak üzere, bazı vakalarda derinin alttaki damarların görünecek kadar yarı
saydam hale geldiği de gözlenmiştir.
De Barsy sendromuna sahip olan bebeklerde ayırt edici
yüzsel hatlara sahip olabilirler. Ayırt edici özellikler çoğunlukla, olağandışı
büyük ve öne doğru çıkık bir alın (frontal çıkıntı), ince dudaklar, geniş
aralıklı gözler (hipertelorism), yukarı bakan küçük bir burun ve kusurlu büyük
kulaklar içerir. Bu hastalığa yakalanmış bazı bebeklerde bıngıldağın
(fonticulus) geç kapanması gözlemlenmiştir ve ek olarak bıngıldağın normalden
büyük olduğunda bazılarında rapor edilmiştir. De Barsy sendromuna yakalanmış
bazı bebeklerde kas tonusunun kaybı ve anormal seviyelerde gevşek eklemler de
gözlemlenmiştir. İskeletsel bazı anormalliklerde gözlemlenebilir ve bu
anormallikler sık sık çıkık (dislokasyon) yaşamak, yumruk pozisyonunda sabit
ellere ve içe doğru bükülmüş başparmaklara sahip olmak, batık bir göğüs
kemiğine sahip olmak, narin kemiklere (osteoporoz), düşük mineral yoğunluklu
kemiklere sahip olmak gibi anormallikler içerebilir.
De Barsy Sendromlu hastalarda farklı seviyelerde zihinsel
engellilik gözlemlenebilir. Daha birçok farklı semptomlara farklı hastalarda
gözlemlenmiştir. Rapor edilmiş bahsedilmeyen semptomları (https://hpo.jax.org/app/browse/disease/ORPHA:2962 Human Phenotype Ontology) linki takip ederek İngilizce
bir şekilde inceleyebilirsiniz.
Genetik Görülme Sıklığı
De Barsy sendromu 1968 ilk tanımlandığından beri 50’den
az vaka rapor edilmiştir. Vakaların yanlış tanı ve tanımlanamamasından dolayı
sendromun gerçek sıklığını tahmin etmek oldukça zordur. Ek olarak, De Barsy
sendromu doğuştan veya bebekliğin ilk aşamalarında belirgindir.
Kalıtım Paterni/ Deseni
De Barsy sendromu otozomal çekinik bir hastalıktır.
Genetik hastalığa sahip olup olmadığı
kişiye anne babasından gelen gen kombinasyonlarına bağlıdır. Resesif
genetik hastalıklar, De Barsy sendromu gibi, aynı anormal geni kalıtımla anne
babasından alırsa kişide görülür. Eğer bir kişi kalıtımsal olarak bir normal
gen ve bir hastalık geni gelirse bu kişiye bu hastalık bakımından taşıyıcı kişi
denir. İki taşıyıcı ebeveynin resesif hastalığa yakalanmış çocuğa sahip olma
şansı her hamilelikte %25 ve taşıyıcı bir çocuğa sahip olma şansı ise %50, son
olarak iki genide normal olan çocuğa sahip olma şansı ise %25’dir. Risk şansı
cinsiyete göre değişmemektedir.
Teşhis Yöntemleri ve
Tedaviler
De Barsy sendromunun teşhisi yukarıda bahsedilen
karakteristik semptomların fark edilmesine, detaylı bir hastalık bakımından
aile geçmişine ve özelleştirilmiş testlere bağlıdır. Şüphelenilen kişinin
cerrahi bir şekilde alınıp incelenen deri örneği hastanın kutis laksaya sahip
olup olmadığını hakkında bilgi verilebilir.Kutis laksanın farklı genetik
tipleri arasında belirli bir tanesini teşhis etmek oldukça zorlayıcı olduğu
için özelleştirilmiş moleküler genetik testler teşhisi onaylamada
kullanılabilir.
De Basry sendromunun çaresi olmamasından dolayı tedavi
yöntemleri, her ne kadar vaka sayısı oldukça az olmasından dolayı tedavi
protokol olmasa da, hastaya özgü semptomları azaltma ve ya yok etme üzerine
odaklanılmıştır. Bu özgü semptomları tedavi yöntemlerine örnek olarak
iskeletsel sorunları cerrahi olarak düzeltilmesi, deri semptomları azaltmak
üzere estetik ameliyat verilebilir.
Akantokilonemiyaz, akantokilonemiyaz persantı olarak
bilinen bir parazit yüzünden ortaya çıkan, ve parazitik hastalık gruplardan
biri olan filaryal hastalıkları (mematod diğer adı ile yuvarlak kurtçuk)
grubuna ait az görülen tropik bulaşıcı hastalıktır.
Bu parazit daha çok afrika gibi tropik iklimlerde
yayılım göstermekte olup küçük sineklerin ısırması sonucunda taşınır. (A. coliroides). [1]
Tropikal hastalıklar tropikal
ve subtropikal (tropikal iklimden biraz daha serin olan iklim
kuşağı) bölgelere özgü ya da o bölgelerde sıklıkla karşılaşılan
hastalıklardır.
Ilıman iklimlerde, grip sezonunun görülme sıklığı
tropik iklimlere göre yüksek olsa da, böceklerin populasyonunu kış uykusuna
zorlanarak kontrol altında tuttuğu için böceklerle taşınan hastalıkların
görülme sıklığı düşüktür. Fakat sera gazı etkisi ve atmosferdeki küresel
sıcaklık artışı farklı enlemlerde daha önceden görülmeyen (Amerikanın Güneyi,
Akdeniz bölgesi, vb.) tropikal hastalık ve vektörlerin yayılmasını
etkilemektedir.
Başka bir türün organizması (konakçı) içinde ya da
üzerinde yaşayıp, yaşadığı organizmanın nütrientlerinden (besinlerini/
besleyici öğelerini) ona zarar verip faydalanan organizmalara parazit denir.
Filaryal, filariodea tipi ipliğe benzeyen parazitik
yuvarlak kurtçuklardan bulaşıcı tropikal parazitik bir hastalıktır.
Belirti ve Semptomlar
Akantokilonemiyaz genel olarak başlangıç semptomları
göstermez. Sonraları semptomlar kendini, deride kırmızılaşma ve kaşıntı
(pruritus), karın ve göğüs ağrısı, kas ağrıları (miyalji) ve ödem olarak gösterebilir.
Bunlara ek olarak, karaciğer ve dalakta abnormal büyümeler de görülebilir
(hepatosplenomegali).
Önemli ortak laboratuvar bulguları özelleşmiş
beyaz kan hücrelerindeki (eozinofil) artışı semptomlar arasında göstermektedir.
Genetik Görülme Sıklığı
Akantokilonemiyaza mansonella persantı parazitininde
sebep olduğu hastalıklardandır. M.
persantı genellikle orta Afrikada ve Güney Afrikanın bazı bölgelerinde
bulunur.
Parazit tropikal ormanların bataklıklarla birleştiği
arazilerde çokça bulunmaktadır. Yani tropikal ve subtropikal bölgeler filaryata
yol açan memantos ve parazit solucanların sık bulunduğu ana bölgelerdendir.
Bu yüzden en çok bu bölgelerde yaşayan nüfus
etkilenmektedir. Bu hastalığın görülme sıklığı yaşı ilerleyen insanlarda doğru
orantılı olarak artmaktadır.
Parazit, tropikal ormanların bataklıklarla birleştiği
arazilerde çokça bulunmaktadır. Yani, tropikal ve subtropikal bölgeler filaryaya
yol açan memantos ve parazit solucanların sık bulunduğu ana bölgelerdendir.
Bu hastalığın görülme sıklığı yaşı ilerleyen
insanlarda doğru orantılı olarak artmaktadır.
Teşhis Yöntemleri ve Tedaviler
Hastalık genel olarak hastanan alınan belirli miktardaki
kan örneğinin mikroskop altında incelenmesiyle teşhis edilmektedir. Önemli
ortak laboratuvar bulguları özelleşmiş beyaz kan hücrelerindeki (eozinofil)
artışı semptomlar arasında göstermektedir.
Kanında erken lavral (mikrofilarya) halindeki Akantokilonemiyaza
persantı bulunan Akantokilonemiyaza hastalarınının kanından bu lavral safadaki
persant izole edilebilir. Akantokilonemiyaz antifilarya (filarya karşıtı)
ilaçlar ve daha bir çok geliştirilmekte olan ilaçlar ile tedavi edilebilir. Hastalık
için doktor tarafından yazılan ilaçlar genellikle Ivermektin (Ivermectin) ya da
dietil-karbomazin (diethyl-carbamazine ya da kısaca DEC) olmaktadır. Daha büyük
kurtların vücuttan uzaklaştırılması için ameliyat gerekli olabilir. Ağır bir
şekilde seyir etmeyen Akantokilonemiyaz vakaları tedavi gerektirmez.
Hastalığın Diğer İsimleri
Akantokilonemiyazaya bir çok isim verilmiştir ve ilk
olarak mansonelloz olarak, ozzardi, persant ve sitreptokerka türü içeren herhangi üç parazit türleriyle benzeri
yaşam süreci göstermesinden dolayı, isimlendirilmiştir. Fakat şu an daha çok M. persant olarak kabul edilmektedir.
Akantokilonemiyaza için diğer eş anlamlı kelimeler ise şunlardır;
Buschke-Ollendorff
Sendromu, bağ dokusunun genetik durumudur. Deride kansere yol açmayan yumrular
çıkabilir ve bazen hissedilmesi zor olabilir. Bu hastalığa sahip insanlar, hem
deride hem kemikte belirti ve semptomlara rastlayabilirler. Semptomlar 20
yaşından önce görülmeye başlanabilir.
Genetik Değişiklikler/Etken Faktörler
Bu sendroma LEMD3
genindeki mutasyonlar yol açmaktadır. Bu gen, (kemik morfogenetik protein) BMP
proteini ve (dönüştürme büyüme faktörü beta) TGF-β olan 2 yolla
sinyal vermeyi kontrol eden proteinin üretiminde rol oynar. Bu sinyal verme
yolları çeşitli hücre fonksiyonlarını düzenler ve yeni kemik hücrelerinin büyümesinde
rol alır. LEMD3 genindeki mutasyonlar, üretilen LEMD3 proteinin azalmasına
sebep olur. Bu durum, BMP ve TGF-β ile sinyal vermeyi artırır ve bu da kemik dokusunun oluşumunu artırır.
Böylece fazla kemik yoğunluğuyla veya aşırı kemik büyümesiyle sonuçlanır.
Belirti ve Semptomlar
Kansere yol
açmayan deride yumrular (ilk olarak çocuklukta fark edilebilir), derinin bazı
bölgelerinde kemik yoğunluğunun artması (X-ray ile fark edilebilir ve kol ve
bacak kemiklerinin sonunda görülebilir. Ağrı veya acı yapmaz), eklem ağrısı,
duymada bozukluk, kemik ağrısı.
Genetik Görülme Sıklığı
Buschke-Ollendorff
Sendromu, 20 bin insanda 1 görülmektedir.
Kalıtım Paterni/Deseni
Otozomal
dominant geçişli bir hastalıktır.
Teşhis Yöntemleri ve Tedaviler
Teşhis için
genetik testi yapılabilir. Geçerli bir tedavisi yoktur.
Hastalıkla İlişkili Genler
LEMD3
genindeki mutasyonlar bu sendroma yol açmaktadır.
Hiperferritinemi-katarakt sendromu, kandaki
ferritin (hiperferritinemi) , vücudun dokularında aşırı demir birikimine sebeb
olan bir protein, ile karakterize edilen
bir hastalıktır. Bu proteinin birikmesi
yaşamın erken saatlerinde başlar ve göz merceklerinin bulanıklaşmasına neden
olur (katarakt). Etkilenen bireylerde, genel popülasyonda olduğu gibi, 60
yaşından sonra, genellikle bebeklik döneminde gelişir. Cerrahi olarak
çıkarılmayan katarakt, ilerleyen karartmaya ve görme bulanıklığına neden olur
çünkü bulanık lensler gelen ışığı azaltır ve bozar.
Bu bozukluktaki hiperferritinemi genellikle katarakt dışındaki herhangi bir sağlık sorununa neden olmamakla birlikte, kandaki yüksek ferritin seviyeleri, belli karaciğer hastalıklarının bir işareti ile karıştırılabilir. Bu koşullar vücutta aşırı demirle sonuçlanır ve kan alımıyla tedavi edilebilir. Bununla birlikte, hiperferritinemi-katarakt sendromu olan kişilerde aşırı demir yoktur ve tekrarlanan kan alımıyla birlikte az sayıda kırmızı kan hücrelerine (anemi) yol açan düşük demir seviyeleri gelişir. Bu nedenle, gereksiz tedavileri veya karaciğer biyopsileri gibi istilacı test prosedürlerini önlemek için hiperferritinemi-katarakt sendromunun doğru teşhisi önemlidir.
Hastalık tahmini olarak her 200.000 bireyde
bir görülmektedir.
Hastalığın Diğer Etkenleri
Hiperferritinemi-katarakt sendromu, FTL
genindeki mutasyonlardan kaynaklanır. Bu gen, protein ferritinin bir bölümü
(alt birimi) olan ferritin hafif zincirini yapmak için talimatlar sağlar.
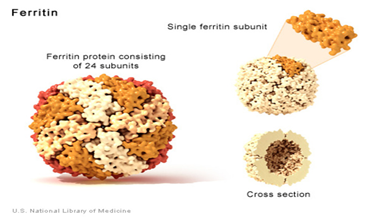
Ferritin, içi boş bir küresel moleküle dönüştürülmüş 24 alt birimden oluşur. 24
alt birim, farklı sayıdaki ferritin hafif zincirinden ve başka bir genden
üretilen ferritin ağır zinciri adı verilen bir alt birimden oluşur. İki alt
birimin oranı farklı dokularda değişir.
Ferritin hücrelerde demir depolar ve
serbest bırakır. Her bir ferritin molekülü, küresel yapısı içerisinde 4.500
kadar demir atomu tutabilir. Bu depolama kapasitesi, ferritinin hücrelerdeki ve
dokulardaki demir miktarını düzenlemesini sağlar.
Hiperferritinemi-katarakt sendromuna neden olan mutasyonlar, demir duyarlı element (IRE) olarak adlandırılan genin bir bölümünde bulunur. IRE normal olarak demir düzenleyici protein (IRP) olarak adlandırılan bir proteine bağlanabilir (bağlanabilir). Bu bağlanma gerçekleştiğinde, çok fazla ferritin hafif zincirinin üretilmesini önlemek için FTL geninin aktivitesi (ifadesi) durdurulur. Bu normalde demir seviyeleri düşük olduğunda meydana gelir, çünkü bu koşullar altında ütüyü depolamak için daha az ferritine ihtiyaç duyulur. FTL geninin IRE segmentindeki mutasyonlar, ferritin üretiminin demir seviyelerine eşleştiği ve aşırı ferritin oluşmasına neden olan mekanizmaya müdahale ederek IRP ile bağlanmasını önler.
Glikojen
depo hastalıkları, enerji sağlamak ve kandaki glikoz değerini korumak için
depolanan glikojenin glikoza dönüştürülemediği
bir hastalık grubudur. Genellikle glikojenin glikoza yıkımını sağlayan
enzimlerden birinin eksikliğinden ortaya çıkar.
Glikojen depo hastalığı tip 1 (GDH 1), sıklığı 1/100 000 olan kalıtsal otozomal
çekinik bir
hastalıktır. GDH 1 belirtileri ve genetik sebeplerinden dolayı ikiye ayrılır:
Glikojen depo hastalığı tip 1A ve glikojen depo hastalığı tip 1B.
Glikojen depo hastalığı tip 1A’da,G6PC genindeki mutasyonlar
glikoz-6-fosfataz (G6Paz) enziminde bir eksikliğe neden olur.Endoplazmik
retikulum lümeninde glukoz 6 fosfat’ın (G6P) glikoz ve fosfata hidrolizini sağlayan glukoz 6 fosfotaz
(G6Paz) enziminin eksikliği glikojen yıkımını engeller. Bu enzim eksikliği
glikoz hidrolizini engeller, aynı zamanda vücuttaki diğer önemli metabolitlerin düzenlenmesi ile sonuçlanarak bu
lipitlerin, özellikle lipidler ve trigliseritler gibi yağların dengesizliğine
veya aşırı birikmesine neden olur. GDH 1A karaciğerde, böbreklerde ve ince bağırsakta glikojen ve yağ birikimine ve bu organlarda
işlev bozukluklarına sebep olur.
Belirti ve Semptomlar
GDH 1A genellikle 3 aylık ve ya 4 yaşları arasındaki
bebeklerde gece boyu uyuma ve yenidoğanlarda yeterince beslenmeme sorunlarıyla
ortaya çıkar. Etkilenen bebeklerde düşük kan şekeri (hipoglisemi) olabilir ve bu da nöbetlere yol açabilir. Ayrıca vücutta laktik asit birikimi (laktik asidoz), ürik asit (hiperürisemi) adı
verilen bir atık ürünün kanda yüksek seviyeleri ve kanda aşırı miktarda yağ
(hiperlipidemi) olabilir. Yaşları ilerleyen çocuklarda ince kol
ve bacak, kısa boy, gövdesel tipte
şişmanlık, ergenliğe geç giriş, karaciğer
ve böbrek büyümesi,
ishal, deride kolesterol birikimi, taş bebek yüzü, kas zayıflığı,
karaciğer büyümesinden dolayı karın
şişkinliği gibi etkiler görülebilir.
Genetik Görülme Sıklığı
GDH 1A otozomal resesif bir hastalık olduğu için aynı
anormal genin her iki ebeveynden de miras kalması gerekir. Eğer birey bir
normal alel gene ve bir de hasta alel gene sahip olursa, bu birey taşıyıcı olur
ve genelde semptomları göstermez. Taşıyıcı
bireyler hastalığın belirtilerini göstermese de çocuklarına
bu geni aktarma riskleri vardır. İki taşıyıcı bireyin çocuklarında
bu hastalığın görülme riski %25’dir, çocukların
taşıyıcı olma riski ise %50’dir. Otozomal bir hastalık olduğu için kadın ve
erkeklerde görülme riski eşittir.
Yakın akraba olan ebeveynlerde akraba olmayan ebeveynlere göre daha
fazla hasta geni taşıma olasılığı vardır.
GDH hastalığının görülme sıklığı
100 000’de 1’dir. GDH 1A ise GDH 1B’den daha yaygındır ve hastaların %80’ini
oluşturur.
Teşhis Yöntemleri ve Tedaviler
Sağlık uzmanları
tipik olarak bir tanı koymak için bir kişinin tıbbi
geçmişine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına
bakar. GSD tip I,
anormal glikoz, laktat, ürik asit, trigliserit ve
kolesterol seviyelerini gösteren laboratuvar testleri ile teşhis edilir. G6PC geni için moleküler genetik testler tanıyı doğrulamak için kullanılabilir.
Moleküler genetik testler ayrıca taşıyıcı testler ve prenatal tanı için kullanılabilir. Moleküler çalışmalar ile erken ve güvenilir tanı
olanağı sağlanmakta, taşıyıcılar belirlenebilmekte ve
genetik danışma verilebilmektedir. Karaciğer biyopsisi GDH 1A için spesifik enzim eksikliğini kanıtlamak için de kullanılabilir. Karaciğer biyopsi örneklerinde artmış glikojen miktarı ve azalmış G6Paz aktivitesi ve/veya G6Paz geninde mutasyon analizi yapılabilir.
GDH 1A normal glikoz seviyelerini korumak, hipoglisemiyi önlemek ve büyüme ve gelişmeyi en üst düzeye çıkarmak için özel bir
diyetle tedavi edilir. Gün boyunca sık sık az porsiyon karbonhidratlar alınmalı ve yaşam boyunca devam etmelidir. Pişmemiş mısır
nişastasının beslenmesi kandaki glikoz seviyelerini iyileştirmek için kullanılır. Kandaki ürik asit seviyesini azaltabilen bir ilaç olan
allopurinol, ergenlik yıllarında gut benzeri artrit semptomlarını kontrol etmek
için yararlı olabilir.
Diyet planı dengeli yapılmalıdır. WHO’nun yaş ve cins için belirlediği ölçütler esas alınmalı ve esansiyel gıdalar mutlaka verilmelidir. Özellikle süt alımı
kısıtlandığından kalsiyum ve vitamin D eksikliği yönünden
dikkatli olunmalıdır. Ayrıca artmış karbonhidrat metabolizması
için yeterli vitamin B1 desteği de yapılmalıdır.
GDH 1A hastaları özel olarak kullanılan
böbrek ve karaciğer ultrasonu ve rutin kan çalışmaları ile en az yılda bir kez
izlenmelidir.
Genetik danışmanlık, etkilenen bireyler ve aileleri için önerilmektedir.
Erken tanı ve diyet tedavisi ile yeterli metabolik kontrolün sağlanması hem komplikasyonların gelişmesini önleyebilmekte hem de hastaların yaşam kalitesini artırmaktadır.
Vici sendromu nadir bir hastalıktır. Erken yaşamda
başlayarak vücudun birçok sistemini etkiler. Beyin, bağışıklık sistmi,kalp,cilt
ve gözde anormallikler ile karakterize bir hastalıktır. Diğer organ ve dokular
ise daha az etkilenmektedir.
Vici sendromunun karakteristik özelliği, beynin sol ve sağ
yarısını (corpus callosum) doğumdan önceki gelişim evrelerinde normalde
oluşturamayan korpus kallozumun ajenezi olarak adlandırılan beyin anomalisidir.
Vici sendromunda beyinde başka anormallikler de meydana gelmektedir, Vici sendromunda beynin, pons (pontine
hypoplasia) olarak bilinen bölgesinin az gelişmiş olması ve sinir hücrelerini
kapsayan ve koruyan bir yağ maddesi olan
miyelin de dahil olmak üzere diğer beyin bölgelerinde anormallikler ortaya çıkabilir.
Beyin gelişimi ile ilgili sorunların yanı sıra, zaman içinde
alışılmadık derecede küçük bir baş büyüklüğü (mikrosefali) ile sonuçlanan beyin
dokusunun bozulması (dejenerasyonu) oluşabilir.
Vici sendromlu
bireylerde beyin sorunları beynin gelişimi olumsuz ve derinden etkiler. Bu
durumdan etkilenen bebeklerde zayıf kas tonusu (hipotoni) vardır. Bu bebeklerin
bir kısmı yuvarlanma eylemini gerçekleştiremez ve yaşlandıkça da bu beceriyi
kaybederler. Neredeyse hiçbirisi düzgün oturamaz, yürüyemez ve bundan etkilenen
çocuklar konuşamaz.
Vici sendromunun bir başka karakteristik özelliği, hayati
tehlike oluşturabilecek tekrarlayan enfeksiyonlara yol açan bozulmuş bağışıklık
işlevidir (bağışıklık eksikliği). En sık
solunum yolu enfeksiyonları görülmektedir. Gastrointestinal ve idrar
yolu enfeksiyonları da sıktır.
Vici sendromlu çocuklarda kardiyomiyopati adı verilen
potansiyel olarak yaşamı tehdit edici kalp rahatsızlığı yaygındır. Zamanla
kötüleşen bu durum, kalbin verimli bir şekilde kan pompalamasını zorlaştırır. Ayrıca
bazı etkilenen çocuklar doğumdan (kalp konjenital) kaynaklanan kalp
bozukluklarına da sahiptir.
Vici sendromunun diğer önemli özellikleri arasında aile
üyelerine ve aynı etnik kökene sahip diğer insanlarda daha açık renkli olan
cilt ve saç (hipopigmentasyon) ve göz merceğinin bozulması (kataraktlar) veya
diğer göz anormalliklerinin görülmesidir.
Vici sendromunun daha
az görülmeyen belirtileri ve semptomları; iç kulakta anormalliklerin neden
olduğu işitme kaybı (duyusal yüz işitme kaybı); ağzın çatısında (yarık damak)
bir açıklık olan veya üst dudakta (yarık
dudak) veya diğer olağandışı yüz bölgelerinde bir açıklık; ve tiroid, karaciğer veya
böbreklerin anormal fonksiyonu gelişebilir. Etkilenen birçok bebek beklenenden
daha yavaş büyür ve kilo alır. Durumun ciddiyeti nedeniyle birçok vici sendromuna yakalanan bireyler 5
yaşının altında yaşamını yitirirler.
Sıklık
Vici sendromu nadir görülen ve sıklığı bilinmeyen bir
hastalıktır. Şu anda yaklaşık 100 kişiye vici sendromu teşhisi konulmuştur.
Nedenler
EPG5 geninteki mutasyon Vici Sendromuna neden olur. EPG5
genindeki mutasyonlar Vici sendromuna neden olur. Bu gen, otofaji adı verilen
hücresel bir süreçte yer alan bir proteini yapmak için talimat sağlar.
Hücreler bu işlemi yıpranmış veya gereksiz hücre parçalarını
geri dönüştürmek veya parçalamak için kullanır. Otofaji ayrıca, enerji ihtiyacı
yüksek olduğunda hücrelerin materyalleri en verimli şekilde kullanmalarına
yardımcı olur. Otofajideki rolüne ek olarak, EPG5 proteini vücudun bakteri ve
virüs gibi yabancı istilacılara karşı bağışıklık tepkisine yardımcı olur.
Vici sendromuna neden olan EPG5 gen mutasyonları, çalışmayan
anormal EPG5 proteinlerinin üretimine yol açar. İşlevsel EPG5 proteini olmadan
yabancı işgalciler, tekrarlayan enfeksiyonlara yol açan bağışıklık
reaksiyonlarına neden olurlar.
Ek olarak, otofaji de bozulmuştur. Araştırmacılar, otofajiye
ilişkin sorunların beyindeki hücrelerin normal gelişimini ve hayatta
kalmalarını, yüksek miktarda enerji gerektiren diğer organ ve dokuları
bozduğunu iddia ediyor; Ancak, bu bozulmanın Vici sendromunun belirti ve
semptomlarına neden olduğu tam olarak açıklanamamaktadır.
Kalıtım Paterni
Bu durum otozomal resesif bir kalıtsal kalıtımla ifade
edilir, yani her bir hücredeki genin her iki kopyası da mutasyonlara sahiptir.
Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona
uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun
belirtilerini ve semptomlarını göstermezler.
Durumun diğer
isimleri
Eksik korpus kallosum katarakt immün yetmezliği,Korpus
kallozum agenezi-katarakt-immün yetmezlik sendromu,Dionisi Vici Sabetta
Gambarara sendromu,Dionisi-Vici-Sabetta-Gambarara sendromu
Yarık dudak / damak, katarakt, hipopigmentasyon ve eksik
korpus kallosum ile immün yetmezlik
Klinik Özellikler
Vici sendromu, bugüne kadar bildirilen ve yaşamın ilk
aylarında ortaya çıkan en yaygın kalıtsal insan multisistemi bozukluklarından
biridir. 5 ana tanısal bulgunun yanı sıra- kallosal agenezis, katarakt,
kardiyomiyopati, hipopigmentasyon ve kombine immün yetmezlik- neredeyse her
türlü organ sisteminin dahil olabileceğini düşündüren çok çeşitli değişken ek
özellikler bildirilmiştir. Üç ek bulgu
(derin gelişimsel gecikme, elde edilen mikrosefali ve belirgin bir şekilde
büyümenin gecikmesi) yakın zamanda ortaya çıkmıştır, ancak spesifik olmamakla
birlikte, 5 ana tanı özelliği ile tutarlı bir şekilde ilişkili olduğu ve tanıyı
desteklediği belirtilmiştir.
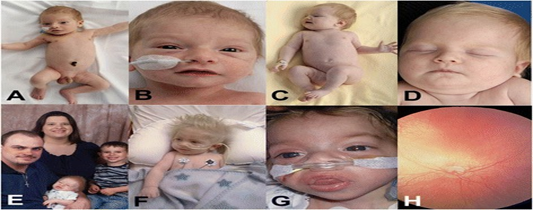
Bireysel hastalarda yapısal konjenital anormallikler ve edinilmiş organ disfonksiyonu (örneğin, doğuştan kalp defekti ve kardiyomiyopati) sık görülmez. Vici sendromundaki tipik bulgular aşağıda ayrıntılı olarak belirtilmiş ve Tablo 1’de özetlenmiştir. Vici sendromunun karakteristik özellikleri Şekil 1’de gösterilmektedir.
Şekil 1. Vici sendromunun klinik özellikleri.
Etnik (A-D, Türkiye
kökenli) ve ailevi (E-F) geçmişe ilişkin hipopigmentasyona dikkat edin. Yüz
özelliklerinin dolgun dudaklarla ve macroglossia’ya benzeyen (lizozomal)
depolanma bozukluklarında kabalaşması bazı çocuklarda görülür (g). Fundoskopide
retinal hipopigmentasyon ve optik atrofiye dair kanıtlar vardır (H). Cullup ve
ark. Nature Genetics 2013; 45 (1): 83–87, izin alınarak çoğaltılmıştır.
Durumun Diğer İsimleri
Eksik korpus kallosum katarakt immün yetmezliği
Korpus kallozum agenezi-katarakt-immün yetmezlik sendromu
Dionisi Vici Sabetta Gambarara sendromu
Dionisi-Vici-Sabetta-Gambarara sendromu
Yarık dudak / damak, katarakt, hipopigmentasyon ve eksik
korpus kallosum ile immün yetmezlik
Kaynakça
1.) Vici
syndrome: a review. Susan Byrne, Carlo Dionisi-Vici, Luke Smith, Mathias
Gautel,2016.11:21
2.) Vici
syndrome,Genetic Home Referance
Website on
Pubmed :PubMed
https://www.ncbi.nlm.nih.gov/pubmed?term=%28Vici+syndrome%5BTIAB%5D%2
9+AND+english%5Bla%5D+AND+human%5Bmh%5D+AND+%22last+2160+days %22%5Bdp%5D
Smith-Magenis sendromu, vücudun birçok bölümünü etkileyen gelişimsel bir hastalıktır. Bu durumun ana özellikleri arasında hafif ila orta dereceli zihinsel engellilik, gecikmiş konuşma ve dil becerileri, belirgin yüz özellikleri, uyku bozuklukları ve davranış sorunları bulunur. Smith-Magenis Sendromu (SMS), genellikle kalıtsal değildir. RAI1 geni; Smith-Magenis
Sendromu ile ilişkili tek gen olup,
sendromun kliniğinden sorumlu olduğu bilinmektedir.
RAI1 geninde oluşan
delesyon veya mutasyonların neredeyse hepsi de novo oluşmaktadır. RAI1 geni
merkezi sinir sistemi ve diğer birçok genin aktivitesinde rol oynar.
Belirti, Semptonlar ve Tanı
Smith-Magenis Sendromu (SMS); Klinik fenotip olgulara göre oldukça farklılık gösterebilmekte ve tanının kesinleşmesi için moleküler analiz gerekmektedir. Tanı erken çocuklukta daha belirsiz, ilerleyen yaşla daha aşikar olan fiziksel, davranışsal ve gelişimsel özelliklerin özgün ve karmaşık paterninin klinik olarak fark edilmesiyle konur.
Moleküler Tanı
Smith-Magenis sendromuna tanı koymak için öncelikle FISH analizi önerilmektedir. Del 17p11.2 FISH analizi hastaların %90’ına tanı imkanı sunmaktadır. Bu analiz ile tanı konulamaması durumunda, hastaların %5-10’unda gözlenen nokta mutasyonlarını belirleyebilen, RAI1 tüm gen sekans analizi önerilmektedir.
Smith-Magenis Sendromu (SMS),
Hastaların %90’ında hastalığın nedeni 17.
kromozomun p11.2 bölgesinin kaybıdır. 25.000’de bir görülen nadir bir hastalıktır. Yaşamın ilk yılında aşırı uyuma ve 12-18 aydan itibaren uyku ritminin bozulması ile kendini kucaklama tarzındaki stereotipik hareket de sendromun karakteristik özellikleri arasındadır. Kendine zarar verme davranışı başını vurma, tırnaklarını çekme, deri koparma, yüzünü tokatlama ve daha karakteristik olarak vücut orifislerine yabancı cisim insersiyonu şeklinde olabilir. Psikomotor ve fiziksel gelişme geriliği, konuşmanın gecikmesi, kaba ses, tipik yüz görünümüne sahip hastalardır. Renal anomaliler, konjenital kalp defektleri iskelet anomalileri, boy kısalığı, uyku düzensizliği, davranış anomalileri, mental retardasyon, ağrı duyusunda azalma, çevre ile uyum problemleri gibi davranış anormalliklerini içeren çoklu konjenital bir anomalidir. Hipotoni en az %90 hastada doğumdan itibaren vardır, daha sonra dil ve motor alanda da gecikme görülür.
Hastaların üçte
birinde konjenital kalp hastalığı, üçte ikisinde işitme kaybı bulunur,
yarısından fazlasında ileri çocukluk yıllarında skolyoz oluşur.
Bu hastalığa yakalanan ortaya çıkan belirtileri toparlayacak olursak; Kare şeklindeki yüz, derin gözler, belirgin alt çene, düz görünen burun, spesifik ağız görünümü, uyku düzeninin bozulması, sık sinir krizi ve patlamalar, genel letarji durumu, tıklama ve tik benzeri istemsiz hareketler, dergi ve kitapları sık sık çevirme, boy kısalığı, skolyoza eğilim, ağrı duyarlılığında azalma, psikomotor gelişimde gecikme, şaşılık, zihinsel gerilik, kalp kusurları, böbrek kusurları, işitme kaybı gibi belirtiler görülür.
Smith Magenis Sendromu Tedavisi
Hastalığı
ortadan kaldırıcı tedavi şuan için
bulunmamaktadır. Temel tedavi mekanizmamız semptomatik ve destekleyicidir.
Konuşma terapisi, mesleki terapi, fiziksel terapi, davranış terapisi, işitme
cihazları, eğitici öğrenme terapisi ve müzik terapisi gibi destekleyici
tedaviler olumlu etki yapmaktadır. Ayrıca bazı antipsikotik ilaçların faydalı
olabildiği bilinmektedir. Erken yaşta ortaya çıkan bu hastalığın tedavi planı
derhal yapılmalıdır. Hemen yapılan tedavi planları neticesinde iyi prognoz elde
edilmektedir. Tanı alan ve tedavi planı yapılan bireyler normal bir insan gibi
hayatını sürdürebilir. Yaşam beklentisi ise normal bireylerle aynıdır.
Kalıtım Paterni
Smith-Magenis
sendromu olan çoğu kişide durum, her hücrede kromozom 17’nin küçük bir
parçasının silinmesinden kaynaklanır. Bu silinme, kromozomun kısa (p) kolunda
p11.2 olarak belirtilen pozisyonda gerçekleşir.
Silinen bölüm, genellikle 3.7 megabaz (Mb) olarak da yazılan yaklaşık
3.7 milyon DNA yapı taşı (baz çifti) içerir. (Bu parçanın ek bir kopyası
Potocki-Lupski sendromu adı verilen ilişkili bir duruma neden olur.) Bazen silme daha büyük veya
daha küçüktür. Silme işlemlerinin tümü, her hücrede bulunan kromozom 17’nin iki
kopyasından birini etkiler. Silinen bölge birden fazla gen içermesine rağmen,
araştırmacılar belirli bir genin, yani RAI1‘in
kaybının, Smith-Magenis sendromunun karakteristik özelliklerinin çoğunun
altındaki sebep olduğuna inanmaktadır. Koşullara neden olduğu bilinen tüm
silinmeler bu geni içerir. RAI1 geni,
diğer genlerin aktivitesini
(ekspresyonunu) düzenlemeye yardımcı
olan bir protein yapmak için talimatlar sağlar. Her ne kadar RAI1 proteini tarafından düzenlenen
genlerin çoğunun tanımlanmamasına rağmen, bu protein uyku-uyanıklık döngüsü
gibi günlük (sirkadiyen) ritimlere dahil olan birkaç genin ekspresyonunu
kontrol ediyor gibi görünmektedir. Araştırmalar, silme işleminin, sirkadiyen
ritimleri etkileyen genlerin ekspresyonunu bozan hücrelerde düşük miktarda RAI1
proteinine yol açtığını göstermektedir. Bu değişiklikler Smith-Magenis
sendromunda meydana gelen uyku bozukluklarını açıklayabilir. RAI1 geninin bir kopyasının
kaybolmasının, bu durumla ilişkili diğer fiziksel, zihinsel ve davranışsal
sorunlara nasıl yol açtığı açık değildir. Smith-Magenis sendromu olan kişilerin
küçük bir yüzdesi, kromozomal silinme yerine RAI1 geninde bir mutasyona sahiptir. Her ne kadar bu bireyler
durumun ana özelliklerinin çoğuna sahip olsalar da, kısa boyları, işitme kaybı
ve kalp veya böbrek anormallikleri olan kişilerden daha az olasıdırlar.
Delesyon (Silinme mutasyonu) olan kişilerde, silinmiş bölgedeki diğer genlerin
kaybının bu ek belirti ve semptomları oluşturması muhtemeldir; bu genlerin rolü
araştırılmaktadır.
Smith-Magenis sendromu genellikle
kalıtsal değildir. Bu durum tipik olarak, bir kromozomal silinme veya üreme
hücrelerinin (yumurta veya sperm) oluşumu sırasında veya erken fetal
gelişiminde meydana gelen bir RAI1 gen mutasyonundan kaynaklanır. Smith-Magenis
sendromu olan çoğu kişinin ailesindeki durumun hiçbir geçmişi yoktur.
Az sayıdaki vakada, Smith-Magenis
sendromu olan insanlar, sadece yumurta hücrelerinde genetik değişimi olan
etkilenmemiş bir anneden silinme ya da mutasyonu miras almışlardır.
Bu fenomene germline mozaikliği denir.
Bu Hastalık İçin Diğer İsimler
17p-sendromu
17p11.2 monozomi
Kromozom 17p delesyon sendromu
Delesyon 17p sendromu
Kısmi monozomi 17p
SMS
Referanslar
Greenberg F, Guzzetta V, De
Oca-Luna RM, et al. Molecular analysis of the SmithMagenis syndrome: a possible
contiguous gene syndrome associated with del(17)(p11.2). Am J Hum Genet 1991;
49: 1207- 1218.
Smith AC, Gropman A.
Smith-Magenis syndrome. In: Cassidy SB, Allanson JE (eds).
Management of Genetic Syndromes (2nd ed). New Jersey: Wiley-Liss,
2005: 507-525
Gropman AL, Duncan WC, Smith
AC. Neurologic and developmental features of the Smith–Magenis syndrome (del
17p11.2). Pediatr Neurol 2006; 34: 337-350
Elsea SH, Williams SR.
Smith-Magenis syndrome: haploinsufficiency of RAI1 results in altered gene
regulation in neurological and metabolic pathways. Expert Rev Mol Med 2011; 13:
e14.
Slager RE, Newton TL, Vlangos
CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith–Magenis
syndrome. Nat Genet 2003; 33: 466-468.
Lurie IW, Lazjuk CL, Ussova I,
Presman EB, Gurevich DB. The Wolf-Hirschhorn syndrome. I. Genetics. Clin Genet
1980; 17: 375-384
Carmona-Mora P, Encina CA,
Canales CP, Cao L, Molina J, Kairath P, Young JI, Walz K. Functional and
cellular characterization of human Retinoic Acid Induced 1 (RAI1) mutations
associated with Smith-Magenis Syndrome. BMC Mol Biol. 2010 Aug 25;11:63. doi:
10.1186/1471-2199-11-63.
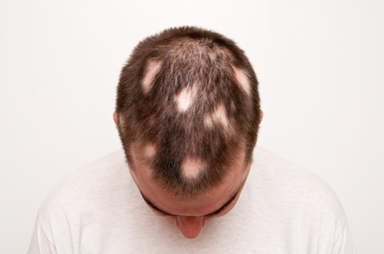
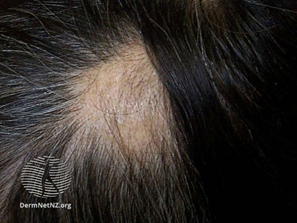
Alopesi areata (halk arasındaki adıyla saçkıran) saç dökülmesiyle karakterize edilmiş, bağışıklık sisteminin saç köklerine saldırıp yok ettiği otoimmün bir hastalıktır. Çoğu vakada saç dökülmesi kafatasında küçük yamalar şeklindedir. Ancak nadir olsa da bazı vakalarda vücudun farklı kısımlarında da etkisini gösterebilir. Hastalık saç derisi kıllarının tamamen kaybolmasına (alopecia totalis) ya da tüm vücut kıllarınının tamamen kaybolmasına (alopecia universalis) kadar ilerleyebilir. Hastalık aktif olsa bile saç kökleri canlı kalır. Bu da saçların tekrardan uzayabileceği anlamına gelir. Saç dökülmesi ve yeniden çıkması döngüsel olabilir.
Saç kaybı genellikle tek semptomdur. Bazı hastalarda yanma hissi ve kaşıntı olabilir. Alopesi areata genellikle bir veya birkaç (1cm-4cm çapında) saç kaybı yaması olarak başlar. Saç kaybının olduğu yamalar düzgün ve yuvarlak bir şekildedir. Şeftali renginde olabilirler. Kel yamaların etrafında ünlem işareti gibi gözüken kıllar görülebilir.
Figür 2: Yama şeklindeki saç kaybı
en belirgin semptomdur.
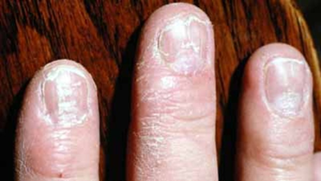
Dökülme en çok saç derisinde görülür. Ayrıca bazı vakalarda dökülme sakal, kaş, kasık, kol veya bacaklarda oluşabilir. Ayrıca bazı vakalarda tırnaklarda çukur oluşumu gibi anormallikler görülebilir.
Alopesi areata her
yaştan kadın ve erkeklerde görülebilir. Ancak genellikle çocukluk yıllarında
başlar. Çocukluk yıllarında başlayan vakalarda
Alopesi universalis gibi ciddi bir tipine dönüşme ihtimali yüksektir. Hastalığın
kadın ve erkeklerde görülme sıklığı eşittir. Literatürde Alopesi areata
prevalansı ile ilgili birkaç tahmin vardır. Medscape Referans tarafından
kullanılan eski kaynaklara göre, genel popülasyondaki prevalans% 0.1-0.2 olarak
tahmin ediliyordu (binde 1 ila 2 aralığında). 2004’teki Ulusal Nadir
Bozukluklar Örgütü’ne (NORD) göre Amerika Birleşik Devletlerinde yaklaşık 2.5 milyon kişi Alopesi
areata hastalığından etkilenmiştir. Bu sayılara göre yaşam boyu riski tahmini
%1-2’dir. Alopesi areata dünya üzerinde görülen en yaygın insan otoimmün
hastalıklarından biridir. Hastalığın görülme sıklığı etnik kökenden
bağımsızdır. Araştırmalar down sendromlu bireylerde ve diğer otoimmün
hastalıklara sahip vakalarda görülme sıklığının daha yüksek olduğunu
göstermişir.
Genetik Değişiklikler/Etken Faktörler
Alopesi areata’nın henüz
kesin nedeni bilinmemektedir. Saç ve ciltte ve bağışıklık sisteminde çalışan
birçok gende meydana gelen değişiklikler de dahil olmak üzere, olası
faktörlerin bir birleşimidir.
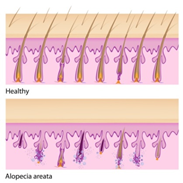
Alopesi areata , otoimmün bozukluklar olarak sınıflandırılan geniş bir bağışıklık sistemi hastalıkları grubundan biridir. Otoimmün bozukluklar, vücudun “yabancı” veya istilacı organizmalara (örneğin antikorlar) karşı doğal savunmasının bilinmeyen sebeplerden dolayı sağlıklı dokuya saldırmaya başlamasından kaynaklanır. Henüz bilinmeyen nedenlerden dolayı, bağışıklık sistemi saç köklerini hedef alır ve saç büyümesini durdurur.
Figür 4: Sağlıklı ve hastalığa sahip
saç köklerinin karşılaştırılması
Alopesi
areata ile ilişkili olan
genlerin çoğu vücudun immün tepkisine katılır. Bunlar, insan lökosit
antijeni (HLA) kompleksi olarak adlandırılan bir gen ailesine
ait birkaç gen içerir. HLA kompleksi, bağışıklık sisteminin vücudun kendi
proteinlerini yabancı istilacılar tarafından yapılan proteinlerden ayırt
etmesine yardımcı olur. Her HLA geninin, her birinin bağışıklık sisteminin
çok çeşitli yabancı proteinlere reaksiyon göstermesine izin veren birçok farklı
varyasyonu vardır. HLA genlerindeki bazı değişiklikler muhtemelen alopesi areata’ya yol açan saç köklerini hedef
alan hatalı immün tepkiye katkıda bulunur . HLA kompleksi dışındaki
bağışıklık sistemi genleri, iltihaplanma ile ilgili birkaç gen gibi,
ayrıca alopesi areata ile de
ilişkilendirilmiştir. Yapılan birkaç GWAS analizi(Genom çapında ilişkilendirme
çalışmaları), birçoğunun immün fonksiyon dahil olduğu bilinen alopesi
areata ile ilişkili 14 genetik lokus tanımlamıştır.
Kalıtım Paterni
Alopecia
areata’nın kalıtım
paterni belirsizdir, Çeşitli kanıtlar, alopesi areata’nın genetik bir
temeli olduğu fikrini desteklemektedir. Genel olarak, durumu geliştirme
riski birinci
derece akrabalar için genel nüfustan daha fazladır. Aile öyküsü olan
yetişkin hastaların prevalansı% 0 ve% 8.6 arasında olduğu tahmin edilmektedir.
Teşhis Yöntemleri ve Tedaviler
Teşhis genellikle saç dökülmesinin
olduğu alanlara odaklanarak yapılır. Ayrıca teşhis için kafa derisi biyopsisi
yapılabilir.
Alopecia areata’nın en
ciddi formu olan Alopecia universalis’in papüler Lezyonlu artişi hastalığıyla
(insan saçsızlık genindeki mutasyondan kaynaklı kalıcı saç kaybı) ayrımının
doğru yapılması tedavi açısından önemlidir.
Şu anda Alopesia areata
için bulunmuş herhangi bir tedavi yöntemi yoktur. Yapılan tedavilerin ana
amacı. Bağışıklık sistemi saldırılarını engellemek ve saçın yeniden büyümesini
teşvik etmek üzerinedir. Bu özellikle hastalığın daha hafif formlarına sahip
kişiler için etkili olabilir. (%50’den az saç dökülmesi) Bu hastalıkta saç
bazen kendi kendine dönebilir. Bazı durumlarda, geri döndükten sonra da
tekrar düşebilir. Bu hastalığın seyri bireyler arasında değişmektedir ve
tahmin edilmesi zordur.
Hafif Alopesi areata için tedaviler
1.İntralezyonel Kortikosteroid Enjeksiyonları
Bu tedavi
yöntemi – alopesi areata için en yaygın uygulanan tedavi şeklidir. – Küçük bir iğne
ile cildin çıplak kısımlarına kortikosteroidler enjekte edilir. Bu
enjeksiyonlar her dört ila altı haftada bir tekrarlanır ve genellikle bir
dermatolog tarafından yapılır.
Kortikosteroidler
(kortizon, prednizon, deksametazon v.b.) vücutta böbrek üstü bezlerinden
salgılanan bir hormon olan kortizole benzer steroid yapıda ilaçlardır.
Avantajları:
Kortikosteroid
enjeksiyonlarından yeni saç büyümesi oluşursa, genellikle dört hafta içinde
görülür.
Dezavantajları:
Kortikosteroid enjeksiyonları, tüm alopesi areata tedavileri gibi, yeni saç dökülmesini engellemez. Bu tür tedavilerle ilgili olarak bildirilen çok az yan etki vardır.
2.lokal olarak Minoksidil uygulanması
Bu tedavi şeklinde, saç derisine, kaşlara ve sakala;
kılların yeniden büyümesine yardımcı olmak için günde bir veya iki kez% 5’lik
bir minoksidil çözeltisi uygulanır.
Avantajları
Bir kişinin saç topikal minoksidil ile tamamen geri
büyürse, tedavi durdurulabilir. Bu ilacın kullanımı kolay kabul edilir ve
minimal yan etkisi vardır.
Dezavantajları
Tek
başlarına kullanıldığında alopesi areata için genellikle etkili değildir, ancak
local kortikosteroid ilaçlarıyla birlikte uygulandığında, bazı insanlar daha iyi
sonuçlar almaktadır.
3.Antralin Kremi veya Merhem
Aynı zamanda sedef hastalığı için de kullanılan, yaygın bir
tedavi şeklidir. Alopesi areata tedavisinde antralin, tüysüz lekelere
günde bir kez uygulanır ve genellikle 30-60 dakika sonra yıkanır.
Avantajları
Antralin uygulamasından yeni saç büyümesi meydana gelirse,
genellikle sekiz ila oniki hafta içinde görülür.
Dezavantajları
Bu ilaç şekli cildi tahriş edebilir ve tedavi edilen ciltte
geçici, solmalara neden olabilir. .
4.Lokal Kortikosteroidler
Alopesi areatada kortikosteroidlerin saç folikülü
çevresindeki iltihabı azalttığı düşünülmektedir. Lokal olarak uygulanan
steroidler; solüsyonlar, losyonlar, köpükler, kremler veya merhemler gibi
farklı şekillerde bulunabilir.
Avantajları
Çalışmalar steroidlerin saç dökülmesinde bir azalma
sağladığını göstermiştir. Ek olarak, yüksek derecede güçlü
kortikosteroidlerin kullanımıyla yaklaşık% 25’lik bir iyileşme
gözlenmiştir. Özellikle alopesi areata olan çocukları tedavi ederken iyi
yardımcı ilaçlar olabilirler.
· Kapsamlı Alopesi areata, Alopesi totalis ve Alopesi universalis için tedaviler
1.ORAL KORTİKOSTEROİDLER
Yoğun saç dökülmesi olan hastalarda hap şeklinde alınan
kortikosteroidler, bazen hastalık aktivitesini baskılamak ve saçları tekrar
büyütmesini sağlamak için reçete edilir.
Avantajları
Saçların uzaması kısa sürede gerçekleşir.
Dezavantajları
Sağlıklı, genç yetişkinlerde genellikle yan etkiler
haplarını tolere edebilir boyuttadır. Ama tedavi amacı ve yan etkileri
hakkında doktorlara danışmak önemlidir. Diğer tedavi yöntemlerinde olduğu gibi,
tedavi sona erdikten sonra oluşan saçlar geri döklebilir.
2.Yerel İmmünoterapi
Yerel immünoterapi, geniş alopesi areata, alopesi totalis
ve alopesi universalis tedavisinde kullanılır. Bu tedavi şekli,
difensipron (DPCP), dinitroklorobenzen (DNCB) veya karik asit dibutil ester
(SADBE) gibi kimyasalların kafa derisine uygulanmasını içerir.
Avantajları
Bu yöntem ile tedavi edilen hastaların yaklaşık% 40’ı,
yaklaşık altı ay tedaviden sonra kafa derisinde saç büyümesi görülür.
Dezavantajları
Tedavi de başarı gözlenen hastalarda saç büyümesinin devam etmesi için tedaviye devam edilmelidir. Kızarıklık, kaşıntı ve uygulama yerlerinde döküntü sık görülen yan etkilerdir.
Ayrıca Morquio Sendromu olarak bilinen Mukopolisakkaridoz
tip 4(MPS 4) başlıca iskeleti etkileyen ilerleyici bir durumdur.Semptomların
kötüleşme oranı etkilenmiş bireylerin arasında çeşitlilik gösterir.
Genellikle MPS 4’ün ilk işaretleri ve semptomları erken
çocukluk döneminde ortaya çıkarlar.Etkilenmiş bireylerde kısa boyu,parantez
bacağı ve kaburgalarda,göğüste,omurgada,kalçalarda ve el bileklerinde
anormallikleri kapsayan çeşitli iskelet anormallikleri gelişir.MPS 4’lü
insanlar gevşek ve çok esnek eklemlere sahiptirler(hipermobil) ama ayrıca
belirli eklemlerde kısıtlı hareket olabilir.Bu durumun karakteristik özelliği
odontoid çıkıntı denen boyunda az gelişmiş(hipoplazi) kanca şekilli
yapıdır.Odontoid çıkıntı boyundaki omurların stabilizasyonuna yardım
eder(servikal vertebra).Odontoid hipoplazi servikal vertebrada hizanın
bozulmasına yönlendirebilir,bu durum baskı yapıp spinal korda zarar verebilir
sonucu paralizi yada ölümdür.Etkilenmiş bazı bireyler tekrarlayan kulak enfeksiyonlarına ve işitme
kaybına sahip olabilir.
MPS 4’lü bireylerde yaşam beklentisi semptomların şiddetine
bağlıdır.Ciddi olarak etkilenmiş bireyler sadece geç çocukluk yada adölesan
döneme kadar sağ kalabilirler.Hastalığın daha ılımlı formlarına sahip olanlar
genellikle yetişkinliğe kadar yaşamalarına rağmen yaşam beklentileri
azalabilir.MPS 4’lü insanlarının asıl ölüm sebepleri spinal kord kompresyonu ve
hava yolu obstrüksiyonudur.
Sebebleri
GALNS ve GLB1 genlerindeki
mutasyonlar MPS 4’e sebep olur.Bu genler glikozaminoglikan(GAGs) adı verilen
büyük şeker moleküllerinin bozulmasını kapsayan enzimlerin üretiminin
talimatlarını sağlarlar.GAGs aslen bu durumun ismini aldığı mukopolisakkaridoz
olarak adlandırılır.MPS 4’e GALNS genindeki mutasyonlar hastalığa sebep
olduğunda MPS 4 Tip A(MPS 4A) olarak
adlandırılır ve GLB1 genindeki mutasyonlar hastalığa sebep olduğunda MPS 4 tip B(MPS 4B) olarak
adlandırılır.Genelde MPS 4’ün 2 tipinde de işaretler ve semptomlar fark
edilebilir olmazlar.
GALNS ve GLB1 genlerindeki
mutasyonlar bu genler tarafından üretilen enzimlerin aktivitesini azaltır yada
tamamen elimine eder.Bu enzimler olmadan
GAGs özellikle lizozomların içinde hücrelerde birikir.Lizozomlar hücrede
parçalayan ve farklı tip diğer moleküllere geri dönüştüren kompartmanlardır.MPS 4 gibi moleküllerin
lizozomlar içinde birikmesi nedeniyle oluşan durumlar lizozomal depo
hastalıklarıdır.MPS 4’te,GAGs çoğu doku ve organlarda,özellikle kemiklerde,
toksik seviyelerde birikir.GAGs’nin birikimi bu hastalıkta kemik
deformitelerine sebep olur.Araştırmacılar MPS 4’ün özelliklerinden GAGs
birikiminin lizozom içerisindeki diğer proteinlerin fonksiyonlarına müdahale
ettiğine ve hücre içerisindeki moleküllerin hareketlerinin bozulmasına yol
açtığına inanmaktadırlar.
Semptomlar
Semptomlar genellikle 1 ila 3
yaşları arasında başlar.Şunları içerirler;
Omurgada dahil kemiklerin
anormal gelişimi
Büyümüş karaciğer
Kalp üfürümü
Kasık fıtığı
Hipermobil eklemler
Boyundan aşağısında sinir
fonksiyonunun kaybı
Özellikle kısa gövdeyle
birlikte kısa boy
Büyük kafa
Geniş boşluklu dişler
Kaba yüz özellikleri
Bulutlu kornea
Dipte genişlemiş
kaburgalarla birlikte kemer şekilli göğüs
Parantez bacak(çarpık
bacak)
Uyku apnesi(ilerleyen
yaşlarda bazı bireylerde görülebilir)
Olası Komplikasyonlar
Komplikasyon olarak şunlar meydana
gelebilir;
Solunum problemleri
Kalp yetmezliği
Spinal kord yaralanması ve
olası paralizi
Görme problemleri
Omurganın anormal eğriliği
ve diğer kemiklerin problemleriyle ilişkili olarak yürüme problemleri
Sıklık
MPS 4’ün kesin prevelansı bilinmemesine rağmen yaklaşık
olarak 200.000 ila 300.000 bireyden 1 bireyde meydana gelmektedir.
Kalıtım Paterni:
Bu durumun kalıtımı,her hücredeki
gendeki iki kopyasında da mutasyona sahip olması anlamına gelen,otozomal
resesif paterndir.Otozomal resesif duruma sahip kişinin ebeveynleri mutasyonlu
genden birer tane kopya taşırlar ama tipik olarak durumun işaret ve
semptomlarını göstermezler.
Hastalığın Engellenmesi
Aile geçmişinde MPS 4 olan ve
çocuk isteyen çiftler için genetik danışmanlık önerilir.Prenatal test uygundur
Muayene ve Testler
Sağlık uzmanı fiziksel muayene
yapacaktır.Fiziksel muayenede şunlara bakılır;
Omurgada anormal eğrilik
Bulutlu kornea
Kalp üfürümü
Kasık fıtığı
Büyümüş karaciğer
Boyundan aşağısında sinir
fonksiyonunun kaybı
Kısa boy(özellikle kısa
gövde)
İdrar testleri genellikle önce yapılır.Bu testler ekstra
mukopolisakkarodizlar gösterebilir ama MPS’nin spesifik formunu
belirleyemezler.Diğer testler şunları içerir;
Kan kültürü
Ekokardiyogram
Genetik test
Duyma Testi
Yarık lamba göz muayenesi
Deri fibroblast kültürü
Uzun
kemiklerin,kaburgaların ve omurganın X-ray’leri
Alt kafatasının ve üst
boynun MRI’ı
Prognoz
Kognitif fonksiyon(açıkça
düşünebilme yeteneği) genellikle MPS 4’lü insanlarda normaldir.
Kemik problemleri büyük sağlık
problemlerine yönlendirebilir.Örneğin,boynun başındaki küçük kemikler kayabilir
ve spinal korda zarar verebilirler,paraliziye sebep olur.Eğer mümkünse bunun
gibi problemleri doğrulayabilmek için cerrahi yapılmalıdır.Kalp problemleri ölüme
yönlendirebilir.
Tedavi
Tip A için,enzimin yerine geçen,
Vimizim denilen ilaç denenebilir.Damardan verilir(IV,intravenöz).Daha fazla
bilgi için uzmanla konuşun.
Enzim replasman(yerine koyma)
terapisi tip B için uygun değildir. İki tip içinde semptomlar meydana geldikçe tedavi edilir.Spinal füzyon boyun kemikleri az gelişmiş insanlarda kalıcı spinal kord yaralanmasını önleyebilir.