Grip, şeker, tansiyon, kardiovasküler rahatsızlıklar gibi yaygın hastalıkların yanı sıra birde nadir hastalıklar vardır; addison hastalığı, anensefali, costello sendromu ve daha bir sürü hastalık bulunmakta. Nadir hastalık adından da anlaşıldığı üzere nüfusa oranla daha az kişide bulunan hastalıklar demektir. Bu hastalıkların yanıtları bilim üzerinden cevaplandırılır. Bu metinde yer alan hastalığın adı “morbid obesity and spermatogenic failure” yani hastalıklı obezite ve sperm azalması. Yetişkin bir bireyin vücudunda olması gereken bir yağ miktarı vardır ve bu vücut kitle endeksi dediğimiz kavram yoluyla ölçülür yani kilonuz boyunuzun karesine bölünmesi sonucunda elde edilen veriler sizin obez olup olmadığınızın kanıtıdır. Dünya Sağlık Örgütünün(WHO) verilerine göre vücut kitle indeksinin sonucu;
• 30 üzeri, obez
• 40 üzeri, morbid obez
• 50 üzeri, süper obez
Obezite en basit anlamıyla vücutta biriken yağ miktarı fazlalığıdır ve bugün obezite halk sağlığı sorunu haline gelmiştir. Yapılan bazı araştırmalar vücut kitle indeksi 40 üzeri olan morbid obezlerde spermatojenik bir azalma gözlemlemişlerdir. Bu araştırmalara ve detaylarına geçmeden önce, morbid obezitenin genetik bir yanının olduğunu, insan ve farelerde “siliyer protein CEP19” kaybının morbid obeziye sebep olduğunu bilmek gerekiyor. (Shalata, 2013) Araştırmalara geri dönecek olursak; shalata ve arkadaşları İsrail asıllı arap aşireti üzerinde bir çalışma yapmışlar ve MO1 sendromu olarak adlandırdıkları otozomal resesif morbid obezitenin etkilerini incelemişler. MO1 lokusu içerisinde kesişen bir mutasyon tespit etmişler; CEP19. (CEP19 bir siliyer proteindir.) CEP19 mutasyon sahibi farelerde hepatik insülin sinyallemesi, bozulmuş yağ oksidasyonu incelemişleridir. (Çalışma Detayları için: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3852924/ )
1992 Yılında Carlsen ve ark. Global olarak sperm yoğunluğunun düşüşünü bildirdiler ve bunun üzerine çalışmalar yaptılar. (ilgili makale: https://www.ncbi.nlm.nih.gov/pubmed/1393072) Bunun üzerine bu çalışmaların yeniden analizi yapıldı ve yayınlandı. (ilgili makale: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1470335/ ) Yeniden analiz sonucunda sperm yoğunluğunda düşüş olduğu yeniden tespit edildi. Sallmén ve arkadaşlarının (Sallmén, 2006) yaptığı çalışma yukarıdaki her iki bilgiyi de içerisinde barındıran bir çalışma olmuştur ve bu çalışma sonucunda kilolu ve obez erkeklerde sperm sayısında azalma olduğu tespit edilmiş. ( ilgili makale: https://www.ncbi.nlm.nih.gov/pubmed/16837825)
Tedavi
• Sağlam bulgular doğrultusunda obezitenin önüne geçecek yöntemler bulunabilir.
• Erkek üreme sağlığı üzerinde çalışmalar yapılabilir.
• İnfertilite tedavisi için tıbbi yollar üretilebilir.
Spermatojenik Azalma Analiz Örneği :

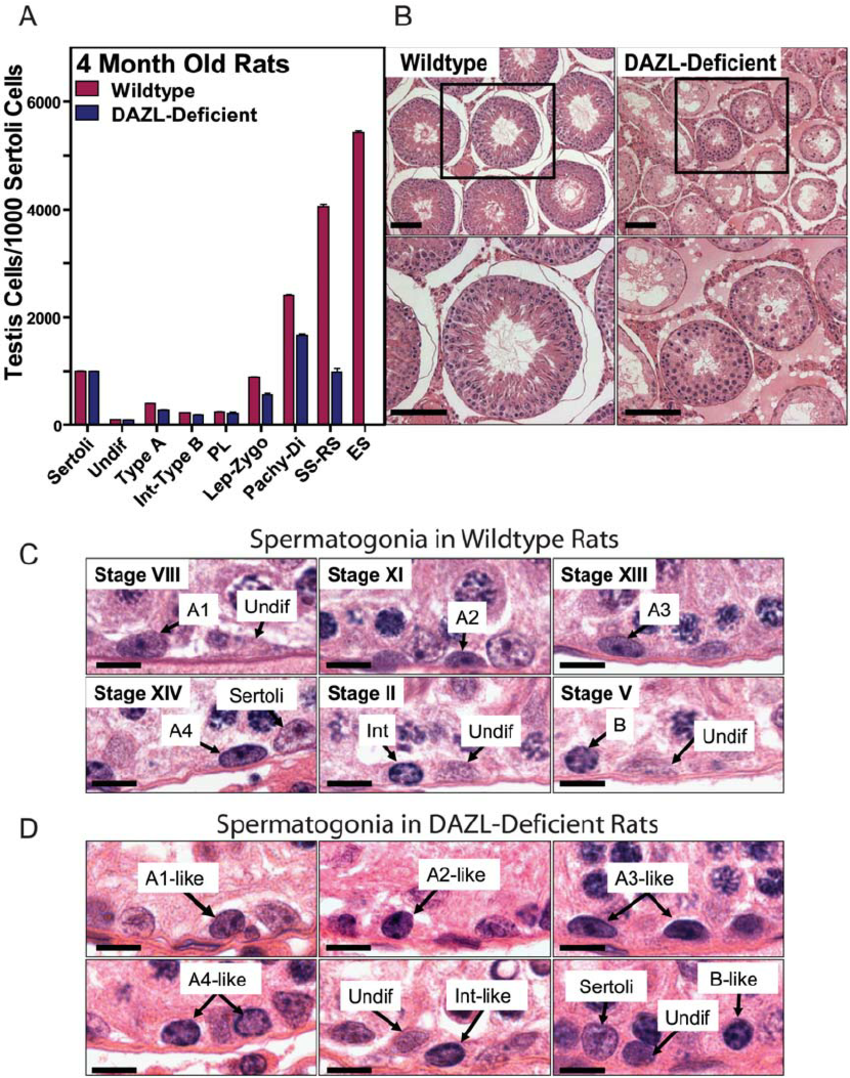
Kaynakça
ACIBADEM HASTANESİ. (tarih yok). ACIBADEM OBEZİTE MERKEZİ: https://www.acibadem.com.tr/obezitemerkezi/detayli-obezite-hesaplama-vucut-kitle-indeksi-hesaplayici/ adresinden alınmıştır
https://www.acibadem.com.tr/obezitemerkezi/detayli-obezite-hesaplama-vucut-kitle-indeksi-hesaplayici/. (tarih yok). Acıbadem hASTANE. adresinden alınmıştır
Sallmén. (2006, eylül 17). Reduced fertility among overweight and obese men.
Shalata, a. (2013). Morbid Obesity Resulting from Inactivation of the Ciliary Protein CEP19 in Humans and Mice.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}