Genel Bilgi
Juvenil miyoklonik epilepsi (JME), miyoklonik jerklerle (kolların veya bacakların hızlı, istem dışı kasılması), jeneralize tonik-klonik nöbetler ve bazen absans nöbetler ile karakterize bir epilepsi sendromudur. JME nöbetleri genellikle insanlar sabah ilk uyandığında ortaya çıkar. Nöbetler uyku eksikliği, aşırı yorgunluk, stres veya alkol tüketimi ile tetiklenebilir. Başlangıç genellikle normalde sağlıklı olan çocuklarda adolesan çağlarında ortaya çıkar. JME’nin nedenleri çok karmaşıktır ve tam olarak anlaşılamamıştır. GABRA1 ve EFHC1 genleri dahil birkaç genin birinde ortaya çıkan mutasyonlar, bu hastalığa neden olabilir veya duyarlılığı artırabilir. Hastaların antikonvülsanlarla ömür boyu tedavileri gerekse de, prognozları genellikle iyidir. (1)
Genetik Değişiklikler/Etken Faktörler
JME’nin kesin nedeni bilinmemektedir. Kafa travması, beyin tümörü veya ensefalit gibi durumlarla ilişkili değildir.
Aile öyküsü ve genetik faktörler JME riskinde güçlü bir rol oynamaktadır. Etkilenen insanların yaklaşık üçte birinin epileptik nöbetleri olan bir akrabası vardır ve birçok ailede spesifik genetik mutasyonlar bulunmuştur. GABRA1 ve EFHC1 genlerindeki ve henüz tanımlanmayan diğer genlerdeki mutasyonlar JME’ye neden olabilir veya duyarlılığı artırabilir.
GABRA1 geni, hücre zarını geçen klorür iyonlarının akması ile ilgili bir proteinin yapılması için talimatlar sağlar. Klorür iyonlarının akışı, hücre içinde sinir hücreleri (nöronlar) arasındaki sinyalleşmeyi engelleyen ve beynin çok fazla sinyalle aşırı yüklenmesini önleyen bir ortam oluşturur. GABRA1 genindeki mutasyonlar beyindeki nöronların aşırı uyarılmasına yol açar ve nöbetlerle ilişkili anormal beyin aktivitesini tetikler.
EFHC1 genindeki mutasyonlar az sayıda insanda JME ile ilişkilendirilmiştir. EFHC1 geni, nöron aktivitesinde de rol oynayan bir proteinin yapılmasına yönelik talimatlar sağlar, fonksiyonu tam olarak anlaşılmamış olmakla birlikte, aynı zamanda nöronların aşırı uyarılmasına ve nöbetlerin tetiklenmesine de yol açabilir.
JME, aşağıdaki belirtilerle ortaya çıkabilir:
- Uyku eksikliği
- Psikolojik stres
- Alkol ve uyuşturucu kullanımı
- İlaç uyumsuzluğu
- Strobe ışıkları gibi titreyen ışıklar
- Regl
- Günün saati – genellikle sabahlar (1)
Belirti ve Semptomlar
JME’nin belirtileri ve semptomları şunlardır:
- Kolların ve bacakların hızlı, istem dışı kasılması olarak tanımlanan ve hastalığın ayırt edici özelliği olan miyoklonik jerkler veya nöbetler vakaların yaklaşık % 17’sinde tek belirti olabilirler; Olguların yaklaşık % 20’sinde nöbetler kümeler halinde görülür, vücudun sadece bir tarafını (tek taraflı) etkiler ve tonik-klonik nöbet öncesi başlar.
- Jeneralize tonik-klonik nöbetler, miyoklonik jerklerin başlamasından birkaç ay sonra ortaya çıkar.
- Absans nöbetleri, genellikle 5 ve 16 yaşlarında ortaya çıkan ilk semptomdur.
- Miyoklonik status epileptikus, JME’yi en çok ilgilendiren problem olarak kabul edilir. Çoklu miyoklonik nöbetler hemen durmadığında, uyku yoksunluğundan ya da kaçırılan nöbetlerden sonra bu durum görülür. (1)
Genetik Görülme Sıklığı
JME, idiyopatik jeneralize epilepsilerin yüzde 25 ile 30’unu ve tüm epilepsi vakalarının yüzde 10’unu oluşturur. Epilepsi riski yüzde 20 olan 20 yaş baz alındığında, JME insidansı 1000 ile 2000’de 1 olarak tahmin edilmektedir.
JME’deki cinsiyet oranının genel olarak eşit olduğu düşünülmektedir, ancak birkaç çalışma kadınlarda 2.9:1’e kadar olan bir üstünlüğü bildirmiştir. (2)
Kalıtım Paterni/Deseni
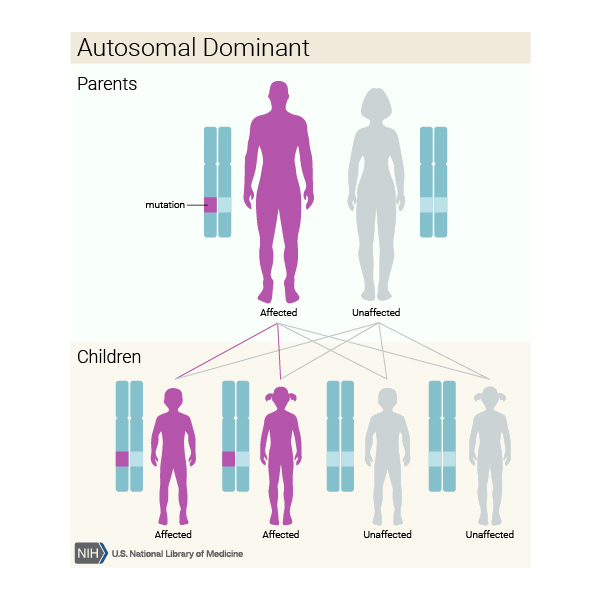
JME’nin kalıtım paterni tam olarak anlaşılamamıştır. Durum GABRA1 genindeki mutasyonlardan kaynaklandığında otozomal dominant bir kalıba kalıtılır, bu da her bir hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olmak için yeterli olduğu anlamına gelir. EFHC1 genindeki mutasyonların neden olduğu juvenil miyoklonik epilepsinin kalıtım paterni bilinmemektedir.
JME epilepsisi ailelerde çalışabilse de, çoğu vakada aile öyküsü bulunmayan kişilerde görülür. (3)
Teşhis Yöntemleri ve Tedaviler
Genetik veya nadir bir hastalık için tanı koymak genellikle zordur. Sağlık uzmanları tanı koymak için genellikle hastanın anamnezine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına bakar.
Alkol kullanımı ve uyku yoksunluğu gibi durumlardan hastayı kaçındırmak faydalı olabilir. Bununla birlikte, anti-konvülsanlarla medikal tedavi genellikle gereklidir ve iyi tolere edilir. Hastaların çoğu tek bir ilaç, en yaygın olarak valproik asit ile iyi kontrol edilebilir. Ayrı ayrı veya kombinasyon halinde kullanılabilecek diğer ilaçlar arasında lamotrijin, levetirasetam, klonazepam ve topiramat bulunur. (1)
Hastalığın Seyri
Nöbetler genellikle nöbet ilaçları ile iyi kontrol edilir ve çalışmalar nöbetlerin yaşamın dördüncü on yılından sonra düzelme eğiliminde olduğunu göstermektedir. Bir kişi nöbet geçirmiyor olsa bile, nöbet ilacı genellikle yüksek tekrarlama riski nedeniyle devam eder (özellikle daha şiddetli formda olanlarda). İnsanların çoğunluğu için ömür boyu tedavi gereklidir. (1)
Hastalıkla İlişkili Genler
- CACNB4
- CLCN2
- EFHC1
- GABRA1
- GABRD (3)
Hastalığın Diğer İsimleri
- adolesan miyoklonik epilepsi
- Janz sendromu
- petit mal, impulsif (3)
Referanslar