Genel Bilgi
Oküler albinizm tip I (OA1) veya X’e bağlı oküler albinizm, oküler albinizmin en yaygın şeklidir. İlk kez 1909’da Nettleship tarafından tanımlanmıştır. Oküler albinizm, X’e bağlı resesif bir genetik hastalık olarak kalıtsaldır. X’e bağlı olduğu için hastalık büyük çoğunlukla erkeklerde görülür ve görme bozuklukları ile karakterize genetik bir hastalıktır. Görme bozuklukları doğumda mevcuttur yani konjenitaldir ve zamanla daha şiddetli hale gelmez. Oküler albinizm, öncelikle gözdeki pigment üretimini etkiler. Sinirlerde retinadan beyne giden anormal bağlantılar, gözlerin birlikte hareket etmesini, izlemesini önler ve derinlik algısını azaltır. Gözler istemsiz bir şekilde ileri geri hareket edebilir.
[1,3]
Belirti ve Semptomlar,
Oküler albinizm öncelikle gözdeki pigment üretimini etkiler. Oküler albinizmde gözlerin ileri geri hareketi (nistagmus), görme bozukluğu, bazı kişilerde yarı saydamlıkla birlikte gelişen iris pigmentinin azalması (hipopigmentasyon), retina pigmentinin azalması, albinotik fundus, maküler hipoplazi, fovea gelişiminin olmaması (foveal hipoplazi) dahil olmak üzere çeşitli görme sorunları ortaya çıkabilir. Hastalarda derinlik algısında azalma meydana gelebilir. Gözlerin birlikte hareketi ve izlemesi azalabilir. Çapraz gözler (şaşılık) ve ışığa duyarlılık (fotofobi) de yaygındır. X’e bağlı konjenital nistagmus 6, oküler albinizm ile birlikte gelişebilen bir allelik hastalıktır. Etkilenen bireyler, görüş netliğini artırmak için genellikle başlarını çevirir veya sallar. Görme bozuklukları doğumda mevcuttur ve zamanla daha şiddetli hale gelmez. Etkilenen bireyler genellikle normal cilt ve saç pigmentasyonuna sahiptir.
İlerleyen zamanlarda yapılan çalışmalar ve araştırmalar ile oküler albinizmin yalnızca gözleri değil melanozomlar (melanin içeren organel) üzerinde de olumsuz bir durum oluşturabileceği gösterilmiştir. X’e bağlı oküler albinizmi olan Kafkaslar ve siyah tenliler ile yapılan histopatolojik bazı çalışmalar ile, hasta erkeklerin ve heterozigot dişilerin, epidermis melanositlerinde ve keratinositlerinde dev melanozomların varlığı belirlenmiştir (O’Donnell ve ark., 1976; O’Donnell ve ark., 1978; Garner ve Jay, 1980). Etkilenen erkekler bazen ciltlerinde hafif bir hipopigmentasyon gösterir ve sıklıkla vücutlarında konjenital hipopigmente maküller (leke, lezyon) ve bölgeler vardır.
X’e bağlı kalıtılar bir hastalık olduğundan, hastalığın sonuçları, semptomları heterozigot veya homozigot olma durumuna göre ve kadın veya erkek olma durumuna göre değişiklik göstermektedir.
Etkilenen erkekler görüş keskinliğinde azalma, yarı saydam irisler, konjenital nistagmus, fotofobi, foveal refleksleri olmayan fundusun hipopigmentasyonu ve yüksek şaşılık insidansına sahiptir. Renk körlüğü etkilenen bazı erkeklerde belirtilmiş olsa da (Forsius and Erkisson,1964), çoğunlukla renk görmede bir sorun meydana gelmemektedir.
X’e bağlı oküler albinizm için heterozigot olan dişiler yani taşıyıcılar, normal görsel fonksiyonlarına rağmen, genellikle funduslarında (göz yuvarlağının arkasındaki alan) karakteristik ve sıklıkla çarpıcı değişikliklere sahiptir. Bu değişiklikler normal veya hiperpigmente retinanın bölgeleri arasına serpiştirilmiş periyotta hipopigmentasyon alanlarından oluşurken, arka kutuplar genellikle pigment epitelinde bir beneklenme gösterir. Normal fundus durumları da heterozigotlukta gösterilmiştir. Aynı zamanda heterozigot dişilerin irisleri sıklıkla yarı saydamlık göstermektedir.

(Şekil 1) [3]
X’e bağlı resesif oküler albinizmde, heterozigot durumda meydana gelen yarı saydam iris gösterilmiştir.
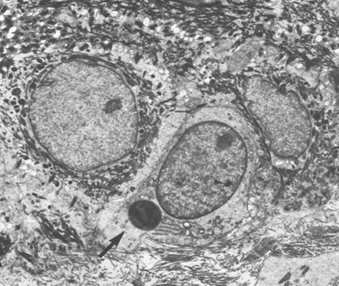
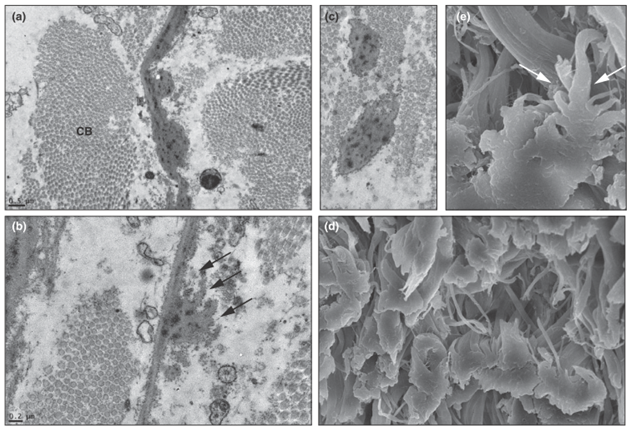
(Şekil 2) [3]
Heterozigot bir bireyin epidermisinin bazal laminası elektron mikroskopu gösterimiştir. Makromelanozom ok ile işaret edilmiştir (x 5.400).
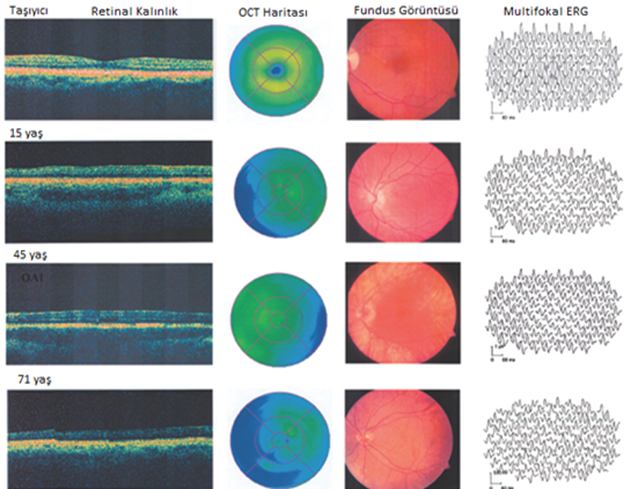
(Şekil 3) [5]
Üç farklı yaştaki erkek hastanın retinal kalınlığı, OCT haritası (optik koherens tomografi), fundus görüntüsü ve multifokal elektroretinografi (ERG) sonuçları verilmiştir. Bir tane de heterozigot taşıyıcı sonuçları da karşılaştırma yapılması açısından eklenmiştir.
[1, 2, 3, 5]
Genetik
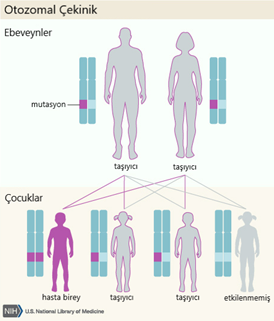
Oküler albinizm, X’e bağlı resesif bir genetik hastalık olarak kalıtsaldır. Hastalığın insidansı 20.000 erkek çocuk doğumunda 1’dir.
404 amino asitlik bir proteini kodlayan, X kromozomu üzerindeki (Xp22) G proteinine bağlı reseptör 143 (GPR143) genindeki mutasyonlar, oküler albinizm ile ilişkilendirilirler. Bu protein, göz ve melanositlerin retinal pigment epitelinde (RPE) eksprese edilir. GPR143, melanin pigment oluşumunun düzenlenmesinde rol oynayan premelanozomal protein MART1 ile etkileşime girer. MART1, GPR143 için bir şaperon proteini olarak işlev görebilir. GPR143’teki mutasyonlar, anormal fibril oluşumuna eşlik eden genişlemiş anormal premelanozomlara neden olur. Premelanozom, pigment hücresindeki melanin pigment üretiminin hücre içi konumudur. Premelanozomlarda melanin pigment sentezinde de bir azalma vardır. Derideki melanozomlarında bozukluklar da mevcuttur, ancak cilt ve saç pigmentinin miktarını azalttığı görülmemektedir.
GPR143’ün, melanin pigment yolunda bir ara metabolit olan L-DOPA (L-3,4-dihidroksifenilalanin) için bir reseptör olduğu düşünülür ve retinada hücre içi sinyalleşmeye katılıyor olabilir. OA1 ile ilişkili mutasyonların çoğu eksik anlatım ile ilgili (missense) mutasyonlardır, fakat anlamsız (nonsense), çerçeve kayması ve splice bölgesi mutasyonları (splice site mutations) da rapor edilmiştir. Bir veya daha fazla ekson dahil olmak üzere birkaç büyük delesyon da bildirilmiştir.
Bazı durumlarda SHROOM2 gibi flanking genler de dahil edilebilir, ancak bu ilave genlerin pigmentasyondaki değişikliklerle ilişkili olmadığı, ancak oküler albinizm tip 1 sendromunda yer alabileceği gösterilmiştir.
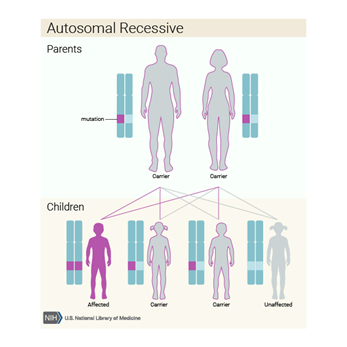
Dişilerin iki X kromozomu vardır, ancak X kromozomlarından biri “kapatılır” ve bu kromozom üzerindeki genlerin çoğu inaktive edilir. X kromozomlarından birinde mevcut bir hastalık geni olan dişiler bu hastalığın taşıyıcılarıdır. Taşıyıcı dişiler genellikle bozukluğun belirtilerini göstermez, çünkü X kromozomunun inaktivasyonu rastgeledir ve genellikle gözdeki hücrelerin yarısında normal X kromozomu aktifleşir ve normal görme ile sonuçlanır.
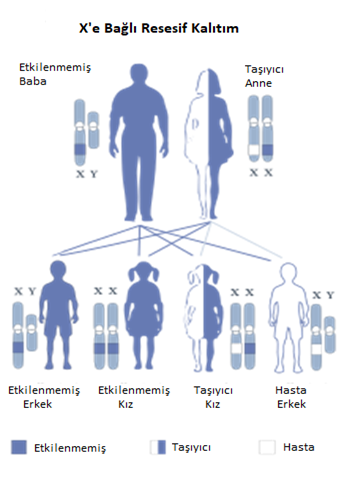
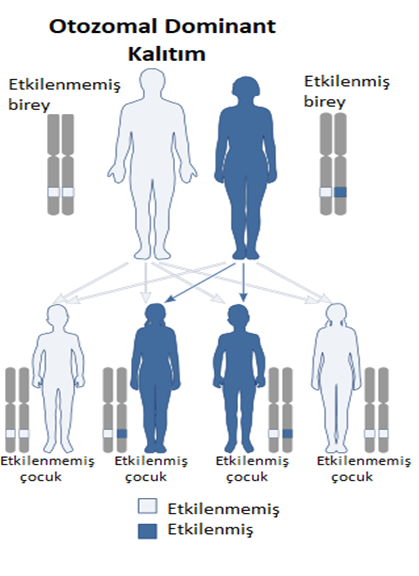
(Şekil 4) [6]
X’e bağlı resesif bir hastalık olduğundan erkekler çoğunlukla hasta olurken kadınlar taşıyıcı olabilirler. Baba hasta, anne hasta ise ise kız çocuk da %100 oranda hasta olur. Kız çocuğa heterozigot bir şekilde geldi ise taşıyıcı olur. Anne taşıyıcı ise erkek çocuklar %50 hasta, hasta ise %100 hasta olur.
Bazı GPR143 mutasyonlarının erkeklerde X’e bağlı konjenital nistagmus ile sonuçlandığına dair kanıtlar vardır.
Tanı
Oküler albinizm tanısı karakteristik göz bulgularına dayanmaktadır.
-Erkekler için:
Oftalmolojik testler yapılır. İnfantil nistagmus, irisin hipopigmentasyonu, oküler fundusun hipopigmentasyonu, foveal hipoplazi. azaltılmış görme keskinliği, anormal optik yol görüntüleri açısından incelemeler yapılır.
İnfatil nistagmus, genellikle yaşamın ilk üç ayında gelişir ve yaşamın ilerleyen döneminde semptom bir düşüş gösterse de nadiren tamamen düzelir. Genellikle yakın mesafeden görüş, uzak mesafeden görüşe göre daha iyidir. İris pigment epitelinin (IPE) hipopigmentasyonu, iris transillüminasyonuna sebep olur. İris pigment epiteli, irisin arka tabakasındadır. Oküler fundusun hipopigmentasyonu, retinal pigment epitelinde (RPE) azalmış pigment konsantrasyonundan kaynaklanır. Foveal hipoplazi, foveal çukurun (umbo) ve halka şeklindeki foveal refleksin azalması veya yokluğu ile karakterizedir. XLOA ilerleyici olmayan bir hastalıktır ve görme keskinliği tipik olarak gençliğin ortasına veya biraz daha sonuna kadar yavaşça ilerler ve daha sonra yaşam boyunca stabil kalır. Çeşitli albinizm formlarında MR görüntülemesi, optik kiazmanın boyut ve konfigürasyonunda normal kontrollere göre farklılıklar bulmuştur.
Dermatolojik olarak, saçta ve deride hipopigmentasyo görülür. Deri renk spektrumu geniştir ve diğer albinizm formlarının tüm spektrumlarını aşar.
-Heterozigot dişiler için:
Genel etnik ve ırka özgü deri ve adneksiyal pigmentasyona bağlı olarak, heterozigot dişiler iris transillüminasyonunu ve vasküler arcade dışında daha dramatik hale gelen retina pigment epitelinin kalın lekeli hipo ve hiperpigmentasyonu gösterebilir. Bazı taşıyıcılar, izole edilmiş hipopigmente cilt lekelerine sahiptir. Nadiren, infantil nistagmus, foveal hipoplazi, azalmış görme keskinliği ve oküler yapıların dağınık hipopigmentasyonunu gösteren heterozigot dişiler belirtilmiştir.
-Hastalığın moleküler tanısı:
Hastalığın moleküler test ile tanısının konulması ve kesin bir şekilde kanıtlanması mümkün olabilmektedir.
GPR143 geni için moleküler genetik test, etkilenen erkeklerin yaklaşık %90’ındaki mutasyonları tespit eder ve tanıyı doğrulamak için kullanılabilir. XLOA tanısı, GPR143’te patojenik bir varyant tanımlaması ile bir probanda konulur. Moleküler test mevcut değilse, cilt biyopsisinde makromelanozomların bulunması da tanıyı koyacaktır.
Moleküler test yaklaşımları, tek gen testini ve bir multigen panelinin kullanımını içermektedir. Tek gen testinde GPR143’ün önce sekans analizi yapılır, ardından patojenik bir değişken bulunmazsa gen hedefli delesyon/dublikasyon analizi yapılır. Multigen panelde ise GPR143 ve diğer ilgili genleri içeren çokgenli bir panel uygulanır. Panele dahil edilen genler ve her bir gen için kullanılan testlerin tanısal duyarlılığı laboratuara göre değişir ve zaman içinde değişmesi muhtemeldir. Panelde kullanılan yöntemler sekans analizi, delesyon/dublikasyon analizi ve/veya sekanslama tabanlı olmayan diğer testleri içerebilir.
Tedavi
Oküler albinizm tanısı konulan bireyler hastalığın gidişatını, kapsamını belirlemek için düzenli olarak muayeneye devam etmektedirler.
Gözlük veya kontakt lensler görüşü büyük ölçüde arttırmaktadır. Güneş gözlüğü kullanımı veya göze siper olucak kenarı olan şapkalar (snapback şapka gibi) güneş hassasiyetinin azaltılmasına yardımcı olabilir. Gözde ışığın kırılması ile ilgili problemler, erkenden uygun gözlük kullanımına başlanmasıyla düzeltilebilir. Nistagmus sebepli anormal baş duruşunu düzeltmek için prizmatik gözlük önerilmektedir. Şaşılık için cerrahi operasyon şart değildir ancak estetik bakımdan istenirse yapılabilmektedir.
Yaşa uygun güneş koruyucu losyonlar ve giysiler için dermatolojik danışma verilmektedir.
Hastalığın Diğer İsimleri
Oküler albinizm tip I (OA1) ,
X’e bağlı oküler albinizm (XLOA),
Nettleship-Falls ocular albinism
Kaynaklar
- JAEGER, C., JAY, B., 1981, X-Linked Ocular Albinism, Hum Genet, 56, 299-304.
- NUSINOWITZ, S., SARRAF, D., 2008, Retinal Function in X-linked Ocular Albinism (OA1), Current Eye Research, 33, 789–803.
{kind=link}