Jeune sendrom öncelikle kemikleri etkileyen nadir bir
durumdur. Yaygın belirti ve semptomlar arasında akciğerlerin büyümesini ve
genişlemesini kısıtlayan küçük bir göğüs ve kısa kaburgalar bulunur ve bu da
genellikle hayatı tehdit eden solunum güçlüklerine neden olur. Diğer
semptomlar kollarda ve bacaklarda alışılmadık şekilli kısaltılmış kemikleri
içerebilir. Kemikler
ve ekstra parmaklar veya ayak parmakları. Bebeklik döneminin nefes alma
zorluklarından kurtulan insanlar daha sonra ciddi böbrek veya kalp problemleri
geliştirebilir. Birçok durumda Jeune sendromunun nedeni
bilinmemektedir; ancak, değişiklikler (mutasyonlar) birkaç farklı genler bu durumdaki bazı ailelerde tanımlanmıştır. Jeune
sendromumiras bir otozomal resesiftavır. Tedavi, her insanda mevcut
olan belirti ve semptomlara dayanır.
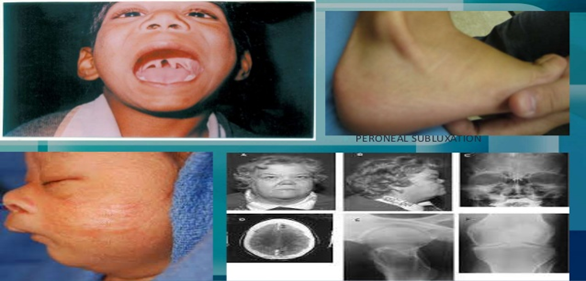
Boğucu torasik distrofi olarak da adlandırılan Jeune sendromu, dar bir
toraks, kısa uzuvlar ve asetabula’nın ‘trident’ yönü ve metafiz
değişikliklerini içeren radyolojik iskelet anormallikleri ile karakterize kısa
bir kaburga displazisidir. Yıllık doğum sıklığı bilinmemektedir ancak 1-5 /
500.000 olduğu tahmin edilmektedir.
Polidaktili olan veya olmayan kısa kaburga torasik displazi (SRTD), dar bir torasik kafes, kısa kaburgalar, kısaltılmış tübüler kemikler ve asetabular çatının ‘trident’ görünümü ile karakterize edilen bir grup otozomal resesif iskelet sililpatisine işaret eder. SRTD, Ellis-van Creveld sendromunu (EVC) ve daha önce Jeune sendrom veya boğucu torakal distrofi (ATD), kısa kaburga-polidaktili sendromu (SRPS) ve Mainzer-Saldino sendromu olarak tanımlanan bozuklukları kapsar.
KROMOZOMAL KONUM
Sitogenetik Yer:
7q36.3, 36.3 konumundaki kromozom 7’nin uzun (q) kolu
Moleküler Yer:
Kromozom 7’de baz çiftleri 158.839.245 ila 158.958.695 (Homo sapiens
Güncellenmiş Ek Açıklama Bülteni 109.20191205, GRCh38.p13)
BU GENİN DİĞER İSİMLERİ
- CFAP163
- DIC6
- FAP163
- SRPS6
- SRTD8
Jeune Sendromun’un Eş anlamları
- boğucu torasik
displazi
- ATD
- torasik-pelvik-falangeal
distrofi
- boğucu
torasik kondrodistrofi
- kondroektodermal
displazi benzeri sendrom
- infantil
torasik distrofi
- Jeune
torasik distrofisi
Etkilenen Popülasyonlar
ATD insidansı 100.000 ila 200.000
canlı doğumda yaklaşık 1’dir. Erkekler ve dişiler, çeşitli etnik veya
ırksal geçmişlere sahip kişiler gibi eşit sayıda etkilenmiş gibi görünmektedir.
Klinik Özellikler
Maroteaux ve Savart (1964) torasik
distrofiyi boğucu olarak tanımladılar ve göğüs kafesi, pelvis ve uzuvlardaki
iskelet değişikliklerinin Ellis-van Creveld sendromunda (EVC; 225500 ) gözlemlenenlere
benzer olduğunu belirttiler . Pirnar ve Neuhauser (1966) etkilenen 3
erkek kardeş bildirmiş ve tırnaklarının displazisi olmadan polidaktili
varlığını belirtmiştir. Erken çocukluk döneminde hayatta kalanlar, kronik
nefrit ( Wahlers, 1966 ) ve bağırsak
malabsorpsiyonu ( Karjoo ve diğerleri, 1973 ) gibi
başka bozukluklar geliştirme eğilimindeydi .
Hanissian
ve diğ. (1967) her biri 2 erkek kardeşi olan 2 aile
bildirmiştir; 1 aile Afrika kökenliydi. Bu yazarlar, Shapira
ve ark. (1965) bu duruma sahipti.
Langer
(1968) , polidaktili olgularda, sadece radyolojik
nedenlerle Ellis-van Creveld sendromundan farklılaşmanın mümkün
olmayabileceğine dikkat çekmiştir. Polidaktili, ATD’nin tutarsız bir
özelliğidir ve mevcut olduğunda genellikle ayakları da etkiler. Aksine,
ellerin polidaktili EVC’de sabit bir özelliktir, ancak ayaklar nadiren
etkilenir. ATD’deki ana viseral anormallik renaldir, oysa EVC’de
kardiyaktır.
Shokeir
(1970) , Norveç ekstraksiyonunun boğucu torasik distrofisi ile
ilişkili 5 etkilenmiş kişiyi tanımlamıştır. Kistik böbrek değişiklikleri
(Potter tip IV) tanımlandı. Kistik lezyonlar böbrek, karaciğer ve
pankreasta ortaya çıkabilir ( Hopper
ve ark., 1979 ; Landing
ve ark., 1980 ).
Finegold
ve diğ. (1971) hipoplastik akciğerli bir olgu ve otopside alveol
sayısında belirgin bir azalma olduğunu bildirmişlerdir.
Oberklaid
ve diğ. (1977) 10 vaka bildirmiştir. Böbrek ve karaciğer
değişiklikleri progresifti ve en az 2 hastada ölüm nedeni böbrek yetmezliği
idi. Dikkate değer bir vaka, hala 15 yaşında ve boy için 25. yüzdelikte
yaşayan bir çocuğun vakasıydı. Küçük bir göğsü vardı, ancak tek radyolojik
bulgu kısa kaburgalardı. 32 yaşında bir hasta Friedman
ve ark. (1975) .
Turkel
ve diğ. (1985) otopside 7 yenidoğan vakasını incelemiş; 2’si
akraba ebeveynlerinden doğan kardeşlerdi. Cüce telaffuz
edilmedi; polidaktili de olan sadece bir bebekte uzuvlar
kısaydı. Enkondral ossifikasyon femur, omur ve kaburga bölümlerinde
düzensizdi. Pulmoner hipoplazi küçük toraks ile ilişkili
idi. Periportal fibroz, safra kanalı proliferasyonu, siroz (1 vakada) ve
değişken pankreatik fibroz da tarif edildi.
Whitley
ve diğ. (1987) , yenidoğan döneminde direkt hiperbilirubinemi ve
hepatik fibroz ile ilişkili karaciğer fonksiyon bozukluğunu tarif
etmişlerdir. Hudgins
ve diğ. (1990) , sirozla ilişkili progresif hepatik disfonksiyonu
olan bu bozukluğa sahip 2 sib tanımlamıştır. Giorgi
ve diğ. (1990) , hafif bir sendrom formuna sahip 2 kız
kardeş tanımlamıştır.
Zack
ve Beighton (1995) akraba çiftli karışık soydan 6
çocuğun 1’inde spondiloenfondromatoz
(bkz. 607944 ) adını
verdiklerini açıkladılar . İlk
olarak 2.5 yaşında görüldüğünde, psödoakondroplazi ( 177170 ) için geçici bir
tanı konulmuştur, ancak daha sonra özellikler spondiloenfondromatozun
radyolojik görünüm tanısına dönüşmüştür. Gez ile, pelvisin 2.5
yaşında konfigürasyonu, boğucu toraks displazisini bir şekilde
düşündürdü. Daha sonra, 13 yaşında çekilen fotoğraflarla belirtildiği
gibi, göğsün belirgin daralması gelişti.
Labrune
ve diğ. (1999) Jeune sendrom ve karaciğer hastalığı
klinik ve laboratuvar bulguları olan 3 çocuk bildirmişlerdir. Karaciğer
tutulumu şiddetliydi ve hepatik fibrozise ve daha sonra portal hipertansiyonlu
biliyer siroza yol açtı. Bir hastada uzamış neonatal kolestaz ilk
tezahürken, diğer 2 hastada fibroz ve hatta siroz geliştiğinde hepatik
lezyonlar geç fark edildi. Ursodeoksikolik asit ile tedavinin, klinik ve
laboratuvar verilerindeki iyileşmeye bağlı olarak hepatik disfonksiyonun
ilerlemesini kontrol ettiği görülmüştür. Yazarlar, serum safra asidi
konsantrasyonu ölçümleri de dahil olmak üzere Jeune sendromlu hastalarda hepatik fonksiyonun düzenli olarak takip edilmesi
gerektiğini önerdiler.
Kajantie
ve diğ. (2001) , yenidoğan semptomları hafif solunum
sıkıntısından asfiksi ve ölüme kadar değişen ATD’li 3 sib tarif
etmişlerdir. Yazarlar, üçüncü trimesterden önce genç siblerin doğum öncesi
tanısında zorluklar olduğunu bildirmişlerdir. Şiddetli etkilenen
hastaların bile yeni yenidoğan yoğun bakım tedavi seçenekleri göz önüne
alındığında uygun bir prognoza sahip olabileceğini önerdiler.
NORMAL İŞLEVLER
WDR60 geni, WD tekrar protein ailesinin bir üyesini
kodlar. WD tekrarları, tipik olarak gl-his ve trp-asp (GH-WD) tarafından
desteklenmiş yaklaşık 40 amino asidin minimal olarak korunmuş bölgeleridir ve
heterotrimerik veya multiprotein komplekslerinin oluşumunu
kolaylaştırabilir. Bu ailenin üyeleri hücre döngüsü ilerlemesi, sinyal
iletimi, apoptoz ve gen regülasyonu dahil olmak üzere çeşitli hücresel
süreçlerde yer alır. Kodlanmış protein dört WD tekrarı içerir ve silya
oluşumunda rol oynayabilir. Bu gendeki mutasyonlar kısa kaburga
polidaktili ve Jeune sendromları ile ilişkilendirilmiştir.
GENETİK DEĞİŞİKLERE İLİŞKİN SAĞLIK KOŞULLARI
Polidaktili olan veya
olmayan kısa kaburga torasik displazi 8 (SRTD8): Kısa kaburga torasik displazi,
bir grup otozomal resesif ciliopati ve daraltılmış torasik kafes, kısa tırtıklı
kemikler ile karakterize asetabular çatının görünümü. Polidaktili değişken
olarak mevcuttur. İskelet dışı tutulum, yarık dudak / damak yanı sıra beyin,
göz, kalp, böbrekler, karaciğer, pankreas, bağırsaklar ve genital organlar gibi
ana organların anomalilerini içerebilir. Hastalığın bazı formları yenidoğan
döneminde ciddi şekilde kısıtlanmış torasik bir kafese ikincil solunum
yetmezliği nedeniyle öldürücüdür, diğerleri ise yaşamla uyumludur. Hastalık
spektrumu Ellis-van Creveld sendromunu, boğucu torasik distrofiyi (Jeune
sendromu), Mainzer-Saldino sendromunu ve kısa kaburga-polidaktili sendrom
BELİRTİLER
Jeune sendromöncelikle kemikleri etkileyen nadir bir durumdur. Bu
durumdan etkilenen insanlar tipik olarak iskelet anormallikleri ile
doğarlar:
- Küçük, dar göğüs
- Kısa kaburgalar
- Kolların ve bacakların kısaltılmış kemikleri
- Alışılmadık şekilli pelvis
- Ekstra parmaklar ve / veya ayak parmakları
Jeune
sendromunun diğer özellikleri şunlardır; yüksek tansiyon, karaciğer hastalığı, pankreas
kistleri, diş anormallikleri ve göz hastalığı görme kaybına yol açabilecek retina
distrofisi denir.
Hastalar tipik olarak yenidoğan döneminde değişken derecelerde solunum
sıkıntısı ve tekrarlayan solunum yolu enfeksiyonları ile başvururlar. Bu
solunum problemleri ATD’nin en ciddi komplikasyonlarıdır ve bu hastalarda
mortalitenin ana nedenidir. Bazı raporlar ATD’li çocukların% 60-80’inin
bebeklik döneminde veya doğumdan sonraki ilk birkaç yıl içinde öldüğünü
göstermektedir. Erken çocukluk döneminde yaşayan hastalar için, solunum
problemleri yaşla birlikte iyileşme eğilimi gösterir, böylece bir hasta alt grubu
ergenlik veya yetişkinliğe yaşayabilir.
Çocuk büyüdükçe ATD’nin diğer komplikasyonları da olabilir: yüksek
tansiyon, böbrek kistleri, pankreas kistleri ve daha az yaygın karaciğer
hastalıkları, diş anormallikleri ve azalmış veya kötüleşen görme (retinal
distrofi).
Etkilenen bireyler, böbrek
yetmezliğine veya arızalara neden olabilecek kronik nefrit (böbrek
rahatsızlığı) geliştirebilir. Kalp anormallikleri ve hava yolunda daralma
da görülebilir.
SEBEPLERİ
Birçok durumda, Jeune’nin sendrom nedeni
bilinmeyen, ancak değişiklikler (mutasyonlar) birkaç
farklı genler( IFT80 , DYNC2H1 , WDR19 , IFT140 ve TTC21B ) bu durumdaki bazı ailelerde tanımlanmıştır. Bu
genlerin tümü,protein içinde bulunan hücresiliya adı verilen ve hücrelerin yüzeyinde mikroskobik, parmak benzeri
çıkıntılar olan yapılar . Kirpilerin gelişimini ve bakımını bozan
mutasyonların Jeune sendromu ile ilişkili belirti ve semptomlara nasıl yol
açtığı net değildir.
11 gendeki mutasyonların ATD’nin bugüne kadar neden olduğu
bulunmuştur. Genler şunlardır: CEP120, CSPP1, DYNC2H1, IFT80,
IFT140, IFT172, TTC21B, WDR19, WDR34, WDR35 ve WDR60 . Etkilenen
bireylerin yüzde 70’inin bu 11 genden birinde mutasyona sahip olduğu tahmin
edilmektedir. Bu genlerdeki mutasyonlar, kemik gelişimini etkileyen
anormal kirpikler proteinlerine yol açar.
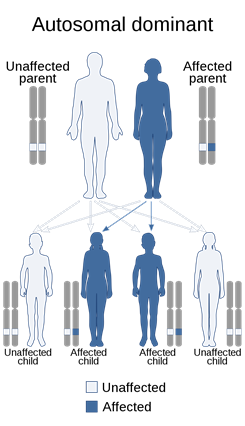
ATD, otozomal resesif genetik bir hastalık olarak kalıtsaldır. Resesif
genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı
anormal geni miras aldığında ortaya çıkar. Bir kişi hastalık için bir
normal gen ve bir gen alırsa, kişi hastalık için bir taşıyıcı olacaktır, ancak
genellikle semptom göstermez. İki taşıyıcı ebeveynin hem değiştirilmiş
geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte%
25’tir. Her hamilelikte ebeveyn gibi taşıyıcı olan bir çocuk sahibi olma
riski% 50’dir. Bir çocuğun her iki ebeveynten normal gen alma şansı%
25’tir. Risk erkekler ve kadınlar için aynıdır.
TEŞHİS
Bazı durumlarda, Jeune tanısı sendromKarakteristik eğer doğumdan önce şüpheli
olabilir belirti ve bulgular mevcut olanultrason. Doğumdan sonra Jeune sendromuRöntgenBulgular. Bazı ailelerde, teşhis
ile doğrulanabilirgenetik test.
- Genetik Test Kayıt (GTR) bu durum için genetik
testler konusunda bilgi sağlar. GTR için hedef kitle sağlık hizmeti
sağlayıcıları ve araştırmacılardır. Genetik test hakkında özel soruları
olan hastalar ve tüketiciler bir sağlık uzmanı veya bir genetik uzmanıyla
görüşmelidir.
- Orphanet , bu durum için
tanısal testler sunan uluslararası laboratuvarları listeler.
TEDAVİ
Tedavi, solunum yolu enfeksiyonlarını yönetmeye ve böbrek ve karaciğer fonksiyonlarını düzenli olarak izlemeye dayanır. Şiddetli solunum yolu enfeksiyonu riski iki yaşından sonra azalır.
Dikey genişletilebilir protez titanyum kaburga (VEPTR), pediyatrik
hastalarda torasik yetmezlik sendromunun (TIS) tedavisi için 2004 yılında FDA
tarafından onaylanmıştır. TIS, göğüs, omurga ve kaburgaların ciddi
deformitelerinin normal nefes almayı ve akciğer gelişimini engellediği
konjenital bir durumdur. VEPTR, omurganın düzeltilmesine ve kaburgaların
düzleştirilmesine yardımcı olan implante edilmiş, genişletilebilir bir
cihazdır, böylece akciğerler büyüyecek ve nefes almak için yeterli hava ile
doldurulabilir. Cihazın uzunluğu hasta büyüdükçe
ayarlanabilir. Spondilotorasik displazi tedavisi için, göğsün her iki
tarafında kaburgalar ayrılır ve göğsün her iki tarafına VEPTR’ler
yerleştirilir. Raynham Mass’ta DePuy Synthes Spine Co. tarafından
üretilmektedir.
ETOLOJİSİ
Sendromun moleküler temeli, her biri bir
intraflagellar taşıma proteinini kodlayan IFT80 (3q25.33), DYNC2H1 (11q22.3),
WDR19 (4p14) ve TTC21B (2q24.3) genlerinin dahil olduğunu gösteren kısmen
açıklanmıştır. Jeune sendromunun ciliopathies grubuna ait
olduğu. Diğer genlerdeki mutasyonlar da hastalığa karışabilir ve
tanımlanmaya devam edebilir.
PROGNOZ
Visseral ilişkili hastalıklara bağlı olarak
prognoz oldukça değişkendir ve ciddi solunum komplikasyonları riski 2 yaşından
sonra azalır.
REFERANSLAR
{kind=link}
{kind=link}
{kind=link}