Juvenil idiyopatik artrit, çocuğun eklemlerinde ağrı ve şişmeye neden olan otoimmün bir durumdur. Bağışıklık sistemi eklemlerin dokularına saldırdığında, ortaya çıkan iltihaplanma eklem hasarına neden olabilir ve bu da hastalığın ağrı ve şişme özelliğine neden olabilir. Juvenil idiyopatik artriti olan bazı çocuklar, alevlenme adı verilen semptomların kötüleştiği bölümlerden etkilenir. Diğer birçok otoimmün hastalık gibi, çocuk idiyopatik artritine genetik ve çevresel faktörlerin bir kombinasyonu neden olur. Juvenil idiyopatik artrit teşhisi için, semptomların 16 yaşından önce başlamış olması gerekir. Durumun teşhisi kan testleri, röntgenler ve diğer benzer durumları dışlayabilir. Tedavi seçenekleri ilaçları ve fizik tedaviyi içerebilir. Juvenil idiyopatik artritin, genetik ve çevresel faktörlerin bir kombinasyonundan kaynaklandığı düşünülmektedir. “İdiyopatik” terimi, bozukluğun spesifik nedeninin bilinmediğini gösterir. Belirtileri ve semptomları eklemlerde ve çevresinde aşırı iltihaplanma nedeniyle ortaya çıkar. Enflamasyon, bağışıklık sistemi mikrobiyal istilacılarla savaşmak ve doku onarımını kolaylaştırmak için sinyal yaratan moleküller ve beyaz kan hücreleri bir yaralanma veya hastalık bölgesine gönderdiğinde ortaya çıkar. Normalde, vücut kendi hücrelerine ve dokularına zarar vermemek için iyileşme tamamlandıktan sonra iltihaplanma tepkisini durdurur. Juvenil idiyopatik artritli kişilerde, özellikle eklem hareketi sırasında enflamatuar yanıt uzar. Bu aşırı enflamatuar yanıtın nedenleri belirsizdir.
Araştırmacılar, çeşitli genlerde jüvenil idiyopatik artrit gelişme riskini etkileyebilecek değişiklikler belirlediler. Bu genlerin bazıları, insan lökosit antijeni (HLA) kompleksi adı verilen bir grup ilişkili proteini yapmak için talimatlar sağlayan bir gen ailesine aittir. HLA kompleksi, bağışıklık sisteminin vücudun kendi proteinlerini yabancı istilacıların (virüsler ve bakteriler gibi) ürettiği proteinlerden ayırt etmesine yardımcı olur. Her HLA geninin birçok farklı normal varyasyonu vardır, bu da her bir kişinin bağışıklık sisteminin çok çeşitli yabancı proteinlere tepki vermesine izin verir. Birkaç HLA geninin bazı normal varyasyonları, çocuk idiyopatik artriti geliştirme riskini ve bir kişinin sahip olabileceği özel durum tipini etkilemektedir.
Diğer bazı genlerdeki normal varyasyonlar da juvenil idiyopatik artrit ile ilişkilendirilmiştir. Bu genlerin çoğunun bağışıklık sistemi işlevinde rol oynadığı düşünülmektedir. Bilinmeyen ek genetik etkiler ve enfeksiyon ve bağışıklık sağlığını etkileyen diğer konular gibi çevresel faktörlerin de bir kişinin bu karmaşık bozukluğu geliştirme şansını etkilemesi muhtemeldir.
Belirti ve Semptomlar
Bu hastalığın fenotipik açıklaması, biyomedikal literatürün bir
analizine dayanır ve İnsan Fenotip Ontolojisi (HPO) terimlerini kullanır.
Fenotipik anormallikler hasta popülasyonunda görülme sıklığı sırasına göre,
daha sonra her bir frekans grubu içindeki alfabetik sıraya göre sunulur. (https://hpo.jax.org/)
Çok Sık
Artralji HP: 0002829
Artrit HP: 0001369
Oto bağışıklık HP: 0002960
Ateş HP: 0001945
Sık
Karın ağrısı HP: 0002027
Anormal eklem morfolojisi HP: 0001367
Tırnakların anormalliği HP: 0001231
Tırnağın anormalliği HP: 0001597
Plevra HP’de anormallik: 0002103
Sakroiliak eklem HP anormalliği: 0100781
Kıkırdak yıkımı HP: 0100773
Genelleştirilmiş hiperkeratoz HP: 0005595
Eklem çıkığı HP: 0001373
Derz sertliği HP: 0001387
Eklem şişmesi HP: 0001386
Malabsorpsiyon HP: 0002024
Mediastinal lenfadenopati HP: 0100721
Tırnak çukurları HP: 0001803
Sedef hastalığı dermatiti HP: 0003765
Ciltte kızarıklık HP: 0000988
Kalınlaşmış cilt HP: 0001072
Üveit HP: 0000554
Nadiren
Hepatomegali HP: 0002240
Perikardiyal efüzyon HP: 0001698
Splenomegali HP: 0001744
Görülme Sıklığı
Kuzey Amerika ve Avrupa’da çocuk idiyopatik artrit sıklığının 10.000 çocukta 4 ila 16 olduğu tahmin edilmektedir. Amerika Birleşik Devletleri’ndeki 1.000 kişiden biri veya yaklaşık 294.000 çocuk etkileniyor. Amerika Birleşik Devletleri’nde en sık görülen çocuk idiyopatik artriti, tüm vakaların yaklaşık yarısını oluşturan oligoartiküler çocuk idiyopatik artrittir. Belirsiz nedenlerden ötürü, kadınlar çocuk idiyopatik artritinden erkeklerden biraz daha sık etkileniyor gibi görünmektedir. Bununla birlikte, entezit ile ilişkili juvenil idiyopatik artritte erkekler kadınlardan daha sık etkilenir. Juvenil idiyopatik artrit insidansı, farklı popülasyonlara ve etnik gruplara göre değişir.
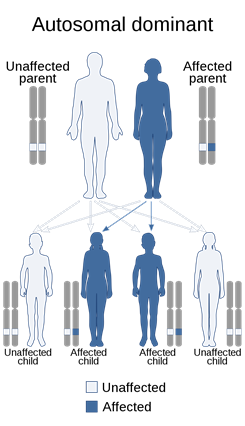
Kalıtım Paterni
Juvenil idiyopatik artrit vakalarının çoğu sporadiktir, bu da ailelerinde bozukluk öyküsü olmayan kişilerde ortaya çıktığı anlamına gelir. Jüvenil idiyopatik artrit vakalarının küçük bir yüzdesinin ailelerde çalıştığı bildirilmiştir, ancak durumun kalıtım paterni belirsizdir. Juvenil idiyopatik artritli bir kişinin kardeşinin, genel popülasyonun yaklaşık 12 katı olan bir durumu geliştirme tahmini riski vardır.
Teşhis Yöntemi
JİA’nın teşhis edilmesi zor olabilir, çünkü bazı çocuklar ilk başta ağrıdan şikayet etmeyebilir ve eklem şişmesi belirgin olmayabilir. Durumu teşhis etmek için kullanılabilecek kan testi yoktur. Romatoid artritli yetişkinlerde tipik olarak pozitif bir romatoid faktör kan testi vardır, ancak JİA’lı çocuklar tipik olarak negatif romatoid faktör kan testine sahiptir. Sonuç olarak, JİA tanısı fiziksel bulgulara, tıbbi öyküye ve diğer tanıların dışlanmasına bağlıdır.
Tipik semptomlar şunları içerir: topallama uyanma sırasında sertlik kol veya bacak kullanma isteksizliği düşük aktivite seviyesi kalıcı ateş eklem şişmesi ince motor aktivitelerinde zorluk
JİA tanısı doğrulanmadan önce enfeksiyonlar, çocukluk çağı kanseri, kemik bozuklukları, Lyme hastalığı ve lupus gibi JIA’ya benzeyen diğer durumlar da göz ardı edilmelidir.
Moleküler genetik testler (veya gen testleri), genetik bir bozukluğa yol açan varyasyonları veya mutasyonları tanımlamak için tek genleri veya kısa DNA uzunluklarını inceler.
Kromozomal genetik testler, genetik bir duruma neden olan bir kromozomun fazladan bir kopyası gibi büyük genetik değişiklikler olup olmadığını görmek için tüm kromozomları veya uzun DNA uzunluklarını analiz eder.
Biyokimyasal genetik testler proteinlerin miktarını veya aktivite seviyesini araştırır; her ikisindeki anormallikler, DNA’da genetik bir bozukluğa neden olan değişiklikleri gösterebilir.
| TEST ADLARI | KOŞULLAR | GENLER | METOTLAR |
| İnflamatuar Bağırsak Hastalığı (IBD) ve İlişkili Bozukluklar, Panel Masif Sekanslama (NGS) 46 Gen | İnflamatuar bağırsak hastalığı 13 (IBD13) Otoimmün lenfoproliferasyonVe sendromu, tip V (ALPS5) Otoenflamasyon, antikor eksikliği ve bağışıklık düzensizliği, plcg2 ile ilişkili (APLAID) Barakat sendromu (HDR) Kandidiyaz, ailesel, 2 (CANDF2) Ailesel kandidiyazis, 6 (CANDF6) | ABCB1ADAM17ATG16L1CYBACYBBDKC1FOXP1FOXP3FUT2 GATA3GUCY2CICOSIKBKGIL10IL10RA IL10RB IL21R IL23R… | Tüm kodlama bölgesinin dizi analizi Yeni Nesil (NGS) / Devasa paralel sıralama (MPS) |
| IL6 TEK GENİ | Serebral arteriyovenöz malformasyon (BAVM) Diabetes mellitus tip 2 (NIDDM) İnflamatuar bağırsak hastalığı 1 (IBD1) Kaposi sarkomu, duyarlılık, laboratuvar tercih edilir: Kaposi sarkomu Romatoid artrit, sistemik çocuk | IL6 | Moleküler genetik DDeletion / çoğaltma analizi Yeni Nesil (NGS) / Devasa paralel sıralama (MPS) Tüm kodlama bölgesinin CSequence analizi Yeni Nesil (NGS) / Devasa paralel sıralama (MPS) |
| MIF GENİ | Rheumatoid arthritis, systemic juvenile | MIF | Moleküler genetik Silme / çoğaltma analizi Yeni Nesil (NGS) / Devasa paralel sıralama (MPS) Tüm kodlama bölgesinin dizi analizi Yeni Nesil (NGS) / Devasa paralel sıralama (MPS) |
Tedaviler
Artritli çocuklar için en iyi bakım, geniş deneyime sahip ve çocuğun ve ailenin karmaşık ihtiyaçlarını en etkili şekilde teşhis edebilen ve yönetebilen bir pediatrik romatoloji ekibi tarafından sağlanır. Çekirdek ekip bir pediatrik romatolog, fizik ve mesleki terapist, sosyal hizmet uzmanı ve hemşire uzmanından oluşabilir. Bu çekirdek ekip, bir çocuğun çocuk doktoru, yetişkin romatologlar, diğer doktorlar (göz doktoru veya ortopedik cerrah gibi) ve diğer sağlık profesyonelleri (diş hekimi, beslenme uzmanı veya psikolog) ile koordine edilebilir ve ayrıca okullara ve ek topluluk kaynaklarına ulaşabilir çocuğun mümkün olan en iyi bakımı almasını sağlar.
Genel tedavi hedefi semptomları kontrol etmek, eklem hasarını önlemek ve işlevi korumaktır. Sadece birkaç eklem olduğunda, herhangi bir ek ilaç verilmeden önce eklem içine bir steroid enjekte edilebilir. Eklem içine enjekte edilen steroidlerin önemli yan etkileri yoktur. Prednizon (Deltasone, Orasone, Prelone, Orapred) gibi oral steroidler belirli durumlarda kullanılabilir, ancak sadece kısa bir süre ve mümkün olan en düşük dozda kullanılabilir. Steroidlerin uzun süreli kullanımı, kilo alımı, zayıf büyüme, osteoporoz, katarakt, avasküler nekroz, hipertansiyon ve enfeksiyon riski gibi yan etkilerle ilişkilidir.
Artrit birçok eklem içerdiğinde veya steroid eklem enjeksiyonlarına cevap vermediğinde, yaygın olarak DMARD olarak adlandırılan hastalık değiştirici ilaçlar eklenir. DMARD’lar arasında metotreksat (Rheumatrex), leflunamid (Arava) ve biyolojik olarak bilinen daha yakın zamanda geliştirilen ilaçlar bulunur. Biyolojiler arasında etanersept (Enbrel), infliksimab (Remicade), adalimumab (Humira), abatacept (Orencia), anakinra (Kineret;), canakinumab (Ilaris), tocilizumab (Actemra) ve rituximab (anti-nekroz faktörleri) bulunur. Rıtuxan). Bu ilaçların her biri, çocuğunuzu tedavi eden pediatrik romatolog ile izlenmesi ve tartışılması gereken yan etkilere neden olabilir. Bu tedavilerin çoğu yetişkinlerde olduğu gibi çocuklarda da onaylanmıştır. Buna ek olarak, araştırmacılar yeni tedaviler geliştiriyorlar.
Hastalıkla İlişkili Genler
- ankyrin tekrar etki alanı 55 – ANKRD55
- CD247 molekülü – CD247
- büyük histouyumluluk kompleksi, sınıf II, DR beta 1 – HLA-DRB1
- interlökin 2 reseptör alt birimi alfa – IL2RA
- interlökin 2 reseptör alt birimi beta – IL2RB
- interlökin 6 – IL6
- 1 – LACC1 içeren lakkaz alanı
- makrofaj göçü önleyici faktör – MIF
- protein tirozin fosfataz reseptör olmayan tip 2 – PTPN2
- protein tirozin fosfataz reseptör olmayan tip 22 – PTPN22
- 4. transkripsiyon sinyal transdüseri ve aktivatörü – STAT4
Hastalığın Diğer İsimleri
- Atrit, genç romatoid
- JİA
- JRA
- Juvenil kronik artrit
- Çocuk RA
- Genç romatoid artrit
- Sistemik çocuk romatoid artrit
Kaynaklar
- https://www.orpha.net/consor/cgi-bin/Disease_Genes_Simple.php?lng=EN&LnkId=720&Typ=Pat&diseaseType=Gen&from=rightMenu
- https://www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Juvenile-Arthritis)
- https://ghr.nlm.nih.gov/
- https://ghr.nlm.nih.gov/condition/juvenile-idiopathic-arthritis#genes
- https://www.orpha.net/consor/cgi-bin/Disease_HPOTerms.php?lng=EN&data_id=720&Typ=Pat&diseaseType=Pat&from=rightMenu
https://rarediseases.info.nih.gov/diseases/3067/juvenile-idiopathic-arthritis, https://ghr.nlm.nih.gov/condition/juvenile-idiopathic-arthritis#genes
{kind=link}