GENEL BİLGİ:
Faktör XI eksikliği , kanın pıhtılaşmasında rol alan faktör XI proteininin bir kıtlığına (eksikliğine) bağlı olarak anormal kanamaya neden olabilecek bir hastalıktır. Bu durum, faktör XI proteininin eksiklik derecesine bağlı olarak kısmi veya şiddetli olarak sınıflandırılır. Bununla birlikte, protein eksikliğinin ciddiyetine bakılmaksızın, çoğu etkilenen birey göreceli olarak hafif kanama problemlerine sahiptir.Faktör XI sendromuna sahip kişilerdeki belirtiler aynı aile içerisinde bile değişebilir.Diğer genetik ve çevresel faktörler hastalığın derecesinin belirlenmesinde etkilidir.
BELİRTİ VE SEMPTOMLAR:
Faktör XI eksikliği vakalarının çoğu , faktör XI proteini yapmak için talimatlar veren F11 genindeki mutasyonlardan kaynaklanır . Bu protein, kan pıhtılarını oluşturan bir dizi kimyasal reaksiyon olan pıhtılaşma kaskadında rol oynar.Yaralanmaya yanıt olarak. Bir yaralanmadan sonra pıhtılar kanamayı durdurmak ve kan damarı onarımını tetiklemek için kan damarlarını kapatır.
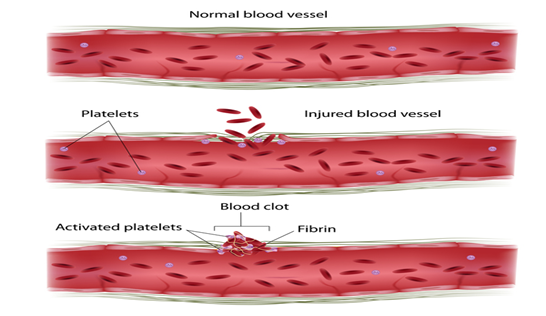
F11 genindeki mutasyonlar , fonksiyonel faktör XI’nin eksikliğine (eksikliğine) neden olur. Bu eksiklik pıhtılaşma kademesini bozar, kanın pıhtılaşma sürecini yavaşlatır ve bu hastalıkla ilişkili kanama sorunlarına yol açar. Kalan fonksiyonel faktör XI miktarı, özel mutasyona ve F11 geninin bir veya her iki kopyasının, her hücrede mutasyona sahip olup olmamasına bağlı olarak değişir . Bununla birlikte, etkilenen bireylerde kanama problemlerinin ciddiyeti mutlaka kan dolaşımındaki faktör XI miktarına karşılık gelmez ve aynı aile içinde bile değişebilir. Diğer genetik ve çevresel faktörler muhtemelen bu durumun ciddiyetinin belirlenmesinde rol oynar.
Faktör XI eksikliğinin en sık görülen özelliği , özellikle ağız ve burun içini (travma veya burun boşlukları dahil) travma veya ameliyat sonrası uzun süreli kanamadır.) veya idrar yolu. Kanama ameliyattan sonra tedavi edilmezse, cerrahi alanda konjuge kandan (hematom) oluşan katı şişlikler gelişebilir.
Bu hastalığın diğer belirti ve semptomları sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri), gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın olmayan faktör XI eksikliği onlar ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez.

Bu hastalığın diğer belirti ve semptomları sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri), gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın olmayan faktör XI eksikliği onlar ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez.
Aşağıda bu hastalığı olan kişilerin sahip olabileceği belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan insanlar listelenen tüm belirtilere sahip olmayabilir. Bu tablo düzenli olarak İnsan Fenotip Ontoloji ((Human Phenotype Ontology [HPO]) isimli veri tabanı tarafından güncellenmektedir. ([https://hpo.jax.org/)
| Tıbbi terimler | Diğer isimler | Daha fazla bilgi edin: HPO Kimliği |
| İnsanların% 80 – 99’u bu belirtilere sahiptir | ||
| Diş çekimi sonrası uzun süreli kanama | 0006298 | |
| Uzun süreli parsiyel tromboplastin zamanı | 0003645 | |
| Azaltılmış faktör XI aktivitesi | 0001929 | |
| İnsanların% 30 -% 79’u bu belirtilere sahiptir | ||
| Epistaksis | Burun kanaması [ daha fazlası ] | 0000421 |
| menoraji | Adet sırasında anormal derecede kanama | 0000132 |
| İnsanların% 1 – 4’ünde bu semptomlar var | ||
| Gastrointestinal kanama | Sindirim sistemi kanaması | 0002239 |
| Eklem kanaması | Eklem içinde kanama [ daha fazlası ] | 0005261 |
| Bu belirtilere sahip kişilerin yüzdesi HPO’da bulunmaz | ||
| Anormal kanama | Kanama eğilimi | 0001892 |
| Otozomal dominant miras | 0000006 | |
| Otozomal resesif miras | 0000007 |
GENETİK GÖRÜLME SIKLIĞI:
Faktör XI eksikliğinin dünya genelinde yaklaşık 1 milyon kişiden 1’ini etkilediği tahmin edilmektedir. Orta ve doğu Avrupa (Aşkenazi) Yahudi soyuna sahip kişilerde, bu popülasyondaki 450 kişiden 1’inde meydana gelen şiddetli bozukluk bozukluğu çok daha yaygındır. Araştırmacılar, faktör XI eksikliğinin gerçek prevalansının bildirilenden daha yüksek olabileceğini öne sürüyorlar , çünkü hastalığın hafif vakaları çoğu zaman tıbbi yardıma gelmiyor.
KALITIM PATERNİ /DESENİ:
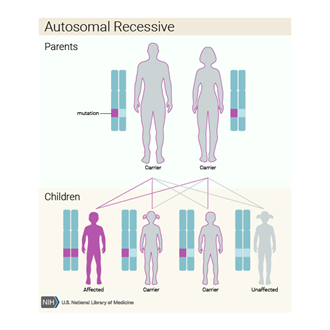
Otozomal resesif paternde ciddi faktör XI eksikliği azaldıbu, her bir hücrede F11 geninin her iki kopyasının da mutasyonlara sahip olduğu anlamına gelir . Bu bireylerin ebeveynlerinin her biri, mutasyona uğramış genin bir kopyasını taşır ve kısmi faktör XI eksikliğine sahiptir ; nadiren durumun ciddi belirtileri ve semptomlarını gösterirler.
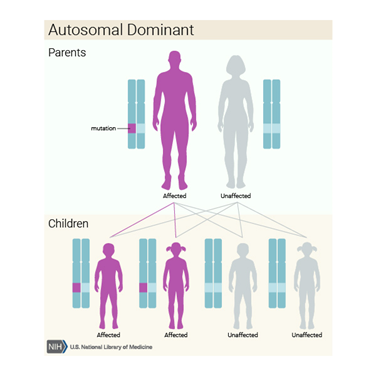
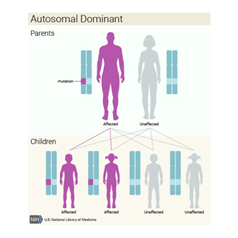
Bazı ailelerde bu durum otozomal dominant paternde kalıtsaldırbu, her hücrede değiştirilmiş F11 geninin bir kopyasının , bozukluğa neden olmak için yeterli olduğu anlamına gelir . Bu gibi durumlarda, etkilenen bir kişinin durumu olan bir ebeveyni vardır.
Kazanılan faktör XI eksikliği formu kalıtsal değildir ve ailelerde çalışmaz.
Teşhis Yöntemleri ve Tedaviler:
Faktör XI eksikliği sendromunun tanısı için 3 önemli belirti şu şekildedir:
1)Ağız ve burun içi travma veya ameliyat sonrası uzun süreli kanama.
2)Sık sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanaması.
3)Bu hastalığa sahip olan kadınlarda ağır veya uzun süreli adet kanaması (menoraji) ve doğumdan sonra uzun süreli kanama.
Ayrıca faktör XI eksikliği sendromu için bazı testler yapılmaktadır. Ancak hastalık tanısı için gerekli değildir.
1)GENETİK TEST:
Genetik test, kromozom, gen veya proteinlerdeki değişiklikleri tanımlayan bir tıbbi test türüdür. Genetik testin sonuçları, şüpheli bir genetik durumu onaylayabilir veya ekarte edebilir veya bir kişinin genetik hastalık geliştirme veya geçme şansını belirlemeye yardımcı olabilir. Şu anda 1000’den fazla genetik test kullanılıyor ve daha fazlası geliştiriliyor.
Genetik test için çeşitli yöntemler kullanılabilir:
Moleküler genetik testler (veya gen testleri), genetik bir bozukluğa yol açan varyasyonları veya mutasyonları tanımlamak için tekli genleri veya kısa DNA uzunluklarını inceler.
Kromozomal genetik testler, genetik bir duruma neden olan bir kromozomun ekstra bir kopyası gibi büyük genetik değişikliklerin olup olmadığını görmek için bütün kromozomları veya uzun DNA uzunluklarını analiz eder.
Biyokimyasal genetik testler, proteinlerin miktarını veya aktivite seviyesini inceler; herhangi birindeki anormallikler, genetik bir bozuklukla sonuçlanan DNA’daki değişiklikleri gösterebilir.
Genetik test isteğe bağlıdır. Testin sınırlamalar ve risklerin yanı sıra faydaları olduğu için, test edilip edilmeyeceği konusundaki karar kişisel ve karmaşık bir karardır. Bir genetikçi veya genetik danışman, testin artıları ve eksileri hakkında bilgi vererek ve testin sosyal ve duygusal yönlerini tartışarak yardımcı olabilir.
2)KISMİ TROMBOPLASTİN ZAMANI (PTT):
Kısmi tromboplastin zamanı (PTT) kanın pıhtılaşmasının ne kadar sürdüğünü gösteren bir kan testidir. Kanama probleminiz olup olmadığını veya kanınızın pıhtılaşmaması durumunda size yardımcı olabilir.
3)PROTROMBİN ZAMANI (PT):
İlgili bir kan testidir.
HASTALIĞIN DİĞER İSİMLERİ:
F11 eksikliği
faktör 11 eksikliği
hemofili C
hemofili C
plazma tromboplastin öncül eksikliği
PTA eksikliği
Rosenthal faktörü eksikliği
Rosenthal sendromu
Rosenthal hastalığı
Faktör XI Eksikliği İçin Yurt Dışındaki Kuruluşlar:
1) Amerika Hemofili Federasyonu
2) Ulusal Hemofili Vakfı
3) Kanada Hemofili Derneği
4) Genetik ve Nadir Hastalıklar (GARD) Bilgi Merkezi
5) Dünya Hemofili Federasyonu
İLGİLİ MAKALELER:
http://www1.wfh.org/publication/files/pdf-1339.pdf
YARARLANILAN KAYNAKLAR:
• https://ghr.nlm.nih.gov/condition/joubert-syndrome
• https://www.omim.org/entry/213300
• https://rarediseases.info.nih.gov/diseases/6802/joubert-syndrome
• https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=1022
• https://rarediseases.org/rare-diseases/joubert-syndrome/


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}