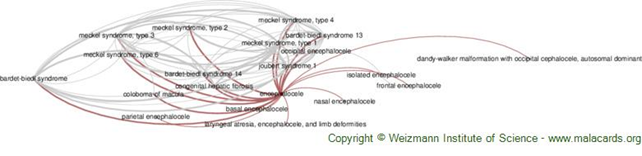
Genel Bilgi
Sandhoff Hastalığı santral sinir sisteminde (beyin ve omurilik) ilerleyici hasarla kendini gösteren, nadir görülen metabolik bir hastalıktır. Lizozomal Heksozaminidaz A ve B enzimlerinin eksikliğine bağlı olarak başta nöronlar (sinir hücreleri) olmak üzere vücuttaki çeşitli dokularda GM2 gangliozid denen lipidler birikir. Bu birikimler nöronlarda işlev bozukluklarına ve nörolojik geriliğe sebep olur. Hastalık hızlı ilerler. Kesin bir tedavisi yoktur, hastalıktan etkilenen bireylere destek tedaviler uygulanır. (1)
Genetik Değişiklikler/Etken Faktörler
5. kromozomun uzun kolunda lokalize olan HEXB adlı gendeki mutasyonlar Sandhoff Hastalığına yol açmaktadır. HEXB geni lizozomal Heksozaminidaz A ve B enzimlerinin üretiminden sorumludur. Bu enzimlerin eksikliğinde bazı moleküllerin yıkımı gerçekleşemez. Bu yıkılamayan moleküllerden olan GM2 gangliozidler beyin ve diğer organlarda birikir ve hastalığın klinik bulguları ortaya çıkar. (2)
Belirti ve Semptomlar
Sandhoff Hastalığının kliniği Heksozaminidaz enzim aktivite düzeylerine göre çeşitlilik gösterir. Hastalığın süt çocuğu formu en sık görülen ve en ağır seyreden formdur. Bebekler normal gelişimlerini sürdürürken 3-6 ay civarında yüksek seslere karşı aşırı irkilme tepkisi göstermeye başlarlar, bunu kazanılmış becerilerdeki kayıp takip eder (dönme, oturma, emekleme vb). Kaslarda güçsüzlük, hipotoni, spastisite ve nöbetlerle seyreden ilerleyici özellikte mental motor gerilik ortaya çıkar. Fizik muayene bulgusu olarak makulada kiraz kırmızısı leke görülebilir fakat bu hastalığa özgü değildir. Karaciğer ve dalakta büyüme, baş çevresinde artış, kemik anomalileri gibi vücuttaki diğer doku ve organları etkileyen klinik tablo görülebilir. Nöbetler kontrol edilse bile hastalığın seyri kötüdür, yutma fonksiyonundaki bozukluk aspirasyona ve tekrarlayan solunum yolu enfeksiyonlarına sebep olur. (3,4)
Sandhoff hastalığının çocukluk, ergenlik ve yetişkinlik dönemlerinde bulgu veren diğer formları çok nadir görülür ve daha iyi seyirlidir. Kas güçsüzlüğü, ataksi (denge bozukluğu), konuşma problemleri gibi çeşitli semptomlarla ortaya çıkabilir. (2)
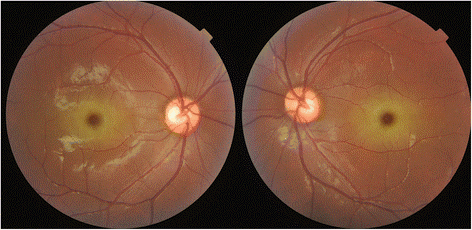
Resim 1: Göz dibi muayenesinde makuladaki kiraz kırmızısı lekeler/Japon Bayrağı görünümü (5)
Genetik Görülme Sıklığı
Sandhoff Hastalığı nadir görülen bir hastalık olduğu için toplumda görülme sıklığıyla ilgili kesin bir sayı vermek güçtür ancak Hacettepe Üniversitesi’nde 1997-2002 yılları arasında yapılan kapsamlı bir çalışmada bu hastalığın Türkiye’deki görülme sıklığının 100.000’de 1 olduğu belirtilmiştir. (6) Bu oran dünyanın farklı bölgelerinde değişiklik gösterir, örneğin hastalık Kanada’nın Saskatchewan bölgesinde yaşayan izole topluluklar gibi bazı etnik gruplarda daha sık görülmektedir. (7)
Kalıtım Paterni
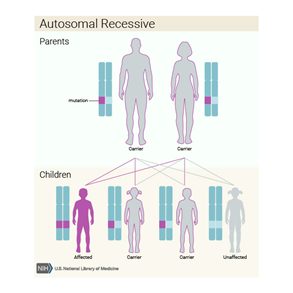
Sandhoff Hastalığı diğer birçok metabolik hastalık gibi otozomal resesif kalıtılır. Bu kalıtım paterninde anne-baba mutasyonlu genin birer kopyasını taşır ama hastalık belirtilerini göstermezler. Kişinin hastalıktan etkilenmesi için mutasyonlu genin her iki kopyasını da taşıması gerekir. Etkilenme oranı kadın ve erkek bireyler için aynıdır. Akraba evliliklerinde daha sık ortaya çıkar.
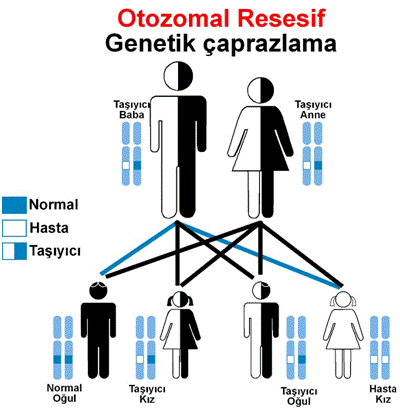
Resim 2: Otozomal resesif kalıtım paterni (8)
Teşhis Yöntemleri ve Tedaviler
Kan örneğinden yapılan enzim çalışmalarında total Heksozaminidaz düşüklüğüyle tanı konur. Tanıyı doğrulamak için gen analizi yapılabilir. (7)
Sandhoff Hastalığına özgü bir tedavi yoktur, semptomatik ve destek tedaviler uygulanır. Sekresyonların temizlenmesi ve havayolu açıklığının sağlanması önemlidir, bunun için medikal cihazlar tercih edilebilir. Hastaya besin ve sıvı desteği verilir, kliniğe göre beslenme tüpü kullanımı gerekebilir. Nöbetlerin kontrolü için antikonvülzan ilaçlar kullanılabilir. Gen terapisi, kök hücre tedavisi ve kemik iliği nakli gibi tedavilere yönelik araştırmalar devam etmektedir. (9,10)
Hastalıkla İlişkili Genler
HEXB geni
Hastalığın Diğer İsimleri
Sandhoff-Jatzkewitz Hastalığı
GM2 Gangliosidosis Tip 2
Heksozaminidaz A ve B Eksikliği
Kaynaklar
1)Muralidharan CG, Tomar RP. Infantile Sandhoff Disease: Unusual presentation. Med J Armed Forces India. 2016;72(Suppl 1):S91–S93. doi:10.1016/j.mjafi.2015.11.008
2) https://ghr.nlm.nih.gov/condition/sandhoff-disease#
3)Yüksel A, Yalçınkaya C, Işlak C, Gündüz E, Seven M. Neuroimaging findings of four patients with Sandhoff disease. Pediatric Neurology. 1999;21(2):562-565. doi:10.1016/s0887-8994(99)00041-7.
4)Jain A, Kohli A, Sachan D. Infantile Sandhoff’s Disease With Peripheral Neuropathy. Pediatric Neurology. 2010;42(6):459-461. doi:10.1016/j.pediatrneurol.2010.02.007.
5)Zou W, Wang X, Tian G. Fundus autofluorescence and optical coherence tomography of a macular cherry-red spot in a case report of sialidosis. BMC Ophthalmology. 2016;16(1). doi:10.1186/s12886-016-0201-9.
6)Özkara HA, Topçu M. Sphingolipidoses in Turkey. Brain and Development. 2004;26(6):363-366. doi:10.1016/j.braindev.2003.09.006.
7)https://rarediseases.org/rare-diseases/sandhoff-disease/
8)https://www.bilim.org/genetik-hastaliklar-ve-akraba-evlilikleri/
9)https://rarediseases.info.nih.gov/diseases/7604/sandhoff-disease#diseaseTreatmentSection
10) https://www.ntsad.org/index.php/infantile-a-juvenile-support/special-equipment