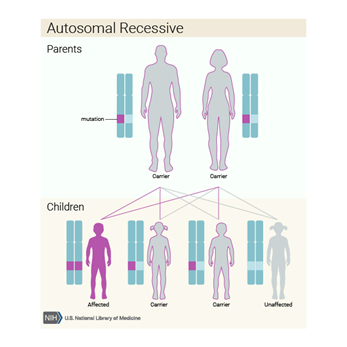
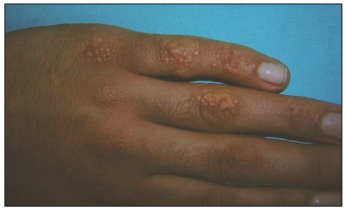
Şilomikron Retansiyon Hastalığı besinlerden yağ, kolesterol
ve bazı vitaminlerin alımının bozulduğu kalıtsal bir hastalıktır. Başlıca
sindirim sistemini ve sinir sistemini etkiler. Bu hastalık bebeklik ya da erken
çocukluk çağında ortaya çıkar. Etkilenen çocuklarda kandaki kolesterol seviyesi
düşüktür. Ayrıca E vitamini eksikliği, büyüme geriliği, sürekli ishal, kötü
kokulu dışkı görülür.
Belirti ve
Semptomlar
Hastaların tümünde kolesterol düşüklüğü ve ishal görülür.
Bunların yanında bireylere göre çeşitlilik gösteren birçok belirti vardır.
Yüksek seviyede karaciğer enzimleri, retina hastalıkları, dışkıda yağ fazlalığı
(steatore), şişkinlik, vitamin metabolizması bozuklukları da etkilenen
bireylerde görülen belirtilerdendir. Çocukluk çağının sonlarında sinir
sisteminde bozukluklar da gelişebilir.
Görülme
Sıklığı
Dünya çapında bugüne kadar yaklaşık 55 vaka tanımlanmıştır.
Genetik
Değişiklikler
SAR1B genindeki
mutasyonlar Şilomikron Retansiyon Hastalığına neden olmaktadır. SAR1B geni Sar1b proteinini kodlar. Bu
protein şilomikron denen moleküllerin taşınmasında rol oynar. Şilomikronlar
sindirim sırasında besin öğelerinin emilimini sağlayan ve yağların, yağda
çözünen vitaminlerin ve kolesterolün ince bağırsaktan kana taşınmasını sağlayan
moleküllerdir. SAR1B geninde meydana
gelen mutasyonlar sonucu bu besin maddelerinin emilimi ciddi ölçüde
azalmaktadır.
Kalıtım
Paterni
Bu hastalık otozomal resesif (çekinik) kalıtılır. Yani
hastalığın oluşması için her iki gen kopyasında da mutasyon olması gerekir.
Teşhis
Yöntemleri
Genelde spesifik semptomlar olmadığı için hastalığın teşhisi
zordur. Teşhis, hastada görülen kronik ishal, yağ emilim bozukluğu ve anormal lipit
profiline (normal trigliserit varlığında, yaklaşık %50 düşük kolesterol) göre
konulabilir. Genetik tanı testleri ile SAR1B
genindeki mutasyonun belirlenmesi mümkündür.
Tedavi
Yöntemleri
Tedavi yaklaşımı beslenme ve büyümenin takibine yönelik
olmalıdır. Uygulanacak tedavi, semptomların belirlenmesi ve önlenmesine
odaklanır. E vitamini eksikliğinin kontrolü nörolojik problemlerin ortaya
çıkmaması için ciddi derecede öneme sahiptir. Tedavi, yağda çözünen vitaminler
ve büyük miktarda E vitamini takviyesi içerir. Diğer belirtilerin önlenmesi
için de yağ alımının çok sıkı takibini içeren özel diyetler uygulanmaktadır.
Hastalığın
Diğer İsimleri
CMRD, Anderson Sendromu, Bağırsakta yağ emilim bozukluğu,
Bağırsak hücrelerinde Hipobetalipoproteinemi’yle birlikte apolipoprotein b
benzeri protein birikimi
Genel
Bilgi, Genetik Değişiklikler/ Etken Faktörler
Proksimal
spinal müsküler atrofiler, omurilik ve beyin sapı çekirdeğindeki alt motor
nöronların dejenerasyonu ve kaybından kaynaklanan ilerleyici kas güçsüzlüğü ile
karakterize bir grup nöromüsküler bozukluktur. Hastalığın başlangıç yaşı ve
şiddetine göre dört alt tip tanımlanmıştır: altı aydan önce başlayan en
şiddetli form olan tip 1 (SMA1); 6 ila 18 aylık arasında başlayan tip 2 (SMA2),
çocukluk ve ergenlik arasında başlayan tip 3 (SMA3) ve yetişkin başlangıcında
en az şiddetli olan tip 4 (SMA4). Tüm tipler, özellikle alt ekstremite ve
solunum kaslarını etkileyen kas zayıflığı ve değişen şiddette atrofi ile
karakterizedir. Zayıflık neredeyse her zaman simetrik ve ilerleyicidir.
Skolyoz, kas retraksiyonları ve eklem kontraktürleri ortaya çıkabilir. Kabızlık
ve gastroözofageal reflü sık görülür.
Hastalık
belirgin olarak SMN proteini sentezleyen SMN1 ve SMN2 geninde gerçekleşen
mutasyonlardan kaynaklanır ve SMN proteini üretemezler. Bunun sonucunda motor
nöron sinirleri beslenemez hale gelir ve istemli kasların çalışmasında sorunlar
meydana gelmektedir.
Belirti
ve Semptomlar
Kişiden kişiye
değişebileceği gibi genelde görülen belirtiler aşağıdaki gibidir.
Kas güçsüzlüğü,
Zihinsel gerilik
Tendon reflekslerinde gerileme
Mikrosefali (baş ve çevresinin
normalden küçük olması durumu)
Alt ekstremite amyotrofisi
(çizgili kas telinin yok olmasıyla sonuçlanabilen hacim azalması)
Skolyoz
Hemiparezi/ kısmi felç
Solunum güçlüğü
EMG anormalliği
–elektromiyografi (EMG) kas hastalığının, hasar bulunan sinirin tespitinde kullanılan
görüntüleme yöntemi
Kabızlık, gasroözofageal reflü
Genetik
Görülme Sıklığı
Her 30.000 kişiden 1’inde görülüyordur.
Kalıtım
Paterni/ Deseni
Otozomal
resesif olarak kalıtılır. Bu sebeple hasta bireyin biyolojik anne ve babasında
mutasyona uğramış ilgili genin bir kopyası taşınıyor demektir. Taşıyıcı olan
ebeveynler hasta değillerdir veya herhangi bir belirti göstermezler.
Teşhis
Yöntemleri ve Tedaviler
Teşhis ve tanı
hastanın tıbbi özgeçmişi ve muayeneye dayanmaktadır. Aynı şekilde yapılan
genetik testlerle doğrulanabilmektedir. Elektromiyografi (EMG) kas
hastalığının, hasar bulunan sinirin tespitinde kullanılan görüntüleme yöntemi
ve kas biyopsisi yapılabilmektedir. Ayırıcı teşhis arasında amiyotrofik lateral
skleroz, konjenital kas distrofileri, konjenital miyopatiler, primer lateral
skleroz, miyastenia gravis ve karbonhidrat metabolizması bozuklukları bulunabilmektedir.
Doğum öncesi tanı ise amniyositlerin veya koryonik villus örneklerinin
moleküler analizi ile mümkündür.
Tedavi olarak
çoğunluğu semptomatik olmak üzere birkaç yöntem bulunmaktadır.
Spiraza,
SMA
hastaları, SMN1 genindeki bir mutasyon nedeniyle hayatta kalma motor nöronu
(SMN) adı verilen bir proteini üretmezler. Spinraza daha az etkili ikinci bir
gen olan SMN2’nin daha fonksiyonel SMN proteini üretme yeteneğini arttırır. Vücuttaki
SMN proteini miktarını arttırarak, Spinraza motor nöron ölümünü geciktirmeye ve
hastalık semptomlarının ilerlemesini yavaşlatmaya yardımcı olabilir.
Zolgensma,
SMN1 geninin sağlıklı bir kopyasını hedef motor nöronlarına vermek için genetik
olarak tasarlanmış bir virüs kullanır. 2 yaş ve altındaki SMA1 hastalarına
uygulanan tek seferlik enjekte edilen bir gen terapisi yöntemidir.
SMA’da özel
olarak araştırılmayan diğer tedaviler hastalık semptomlarını yönetmek veya
komplikasyonları önlemek için de kullanılabilir. Örneğin, kasların sert ve
gergin olduğu spastisite, baklofen, tizanidin veya benzodiazepinler gibi kas
gevşeticiler tarafından hafifletilebilir.
Ayrıca hastada
geilşen semptom ve komplikasyonlar fizik tedavi, nefes tedavisi ve beslenme
destekleri ile bunları engelleme ve geciktirmeye yardımcı olmaktadır.
Hastalıkla
İlişkili Genler
SETX, SMNDC1, FBLN5, KIF1B, SPG7, SMN1,
SMN2, APOE, BICD2, DES, DNAI1, AG genleri genel anlamda hastalıktan sorumlu
genler arasında gösterilmektedir. Her tip SMA’da ilişkili genler veya ilişki
oranları değişmektedir. İlgilendiğiniz SMA tipinin sayfasına bakarak ilişkili
geni bulabilirsiniz.
Kol-Kuşak Kas Distrofisi Tip 2D (LGMD2D), kalça ve omuz bölgelerinin (kol-kuşak bölgeleri)
gönüllü kaslarının israfı (atrofi) ve zayıflığı ile karakterize bir nadir hastalıktır. Kas
zayıflığı ve atrofi ilerleyicidir ve vücudun diğer kaslarını etkileyecek kadar
yayılabilir. Semptomlarının başlangıç yaşı, şiddeti ve ilerlemesi, aynı
ailedeki bireyler arasında bile durumdan duruma değişebilir. Bazı bireylerde
hafif, yavaş ilerleyen bozukluklara sahip olabilirken bazı bireylerde şiddetli
sakatlığa neden olabilecek hızla ilerleyen bir bozukluğa sahip olabilir. Bu
bozukluklar artık genetik ve protein analizi ile ayırt edilebilir.
LGMD2D, alfa-sarkoglikan a proteinin geni olan ve
17q21.33’te lokalize olan (konumlanan) SGCA geninde meydana gelen değişimin
(mutasyonun) neden olduğu otozomal resesif bir genetik durumdur. Genelde bireyde çocukluk veya ergenlik çağında başlar ve ilerleyicidir. Çoğu durumda, LGMD’nin çocukluk çağında
başlaması, ergen veya erişkin başlangıçlı vakalardan daha hızlı ilerleyen daha
ciddi bir bozukluğa neden olur.
Kol-Kuşak Kas Distrofisi
Tip 2D, esas olarak proksimal kasları etkilemesi sonucu yürüme zorluğu,
skapular kanatlanma, baldır psödohipertrofisi ve tiptoe yürüyüş gibi sonuçlara neden olur. Kardiyak
ve solunumsal tutulum nadirdir. Kardiyomiyopati de nadiren
bildirilmiştir.
Genetik
Değişiklikler/Etken Faktörler
Kol-Kuşak Kas Distrofisinde etkili olan gen SGCA
(GKRY) genidir. SGCA geni
sarkoglikan protein kompleksi adı verilen bir protein grubunun alfa bileşeni
(alt-birim) yapmak için talimatlar içerir. Sarkoglikan protein kompleksi,
kas hücrelerini çevreleyen zarda bulunur. Distrofinler ve distrolikanlar
adı verilen proteinlerden oluşan distrofin kompleksine yapışarak (bağlanarak)
stabilize ederek (sağlamlaştırarak) kas dokusunun yapısının korunmasına
yardımcı olur. Büyük distrofin kompleksi, kas liflerini güçlendirir ve
kasları yaralanmaya karşı korur. Her kas hücresinin yapısal çerçevesini
(hücre iskeletini) protein kafes ve hücre dışındaki diğer moleküllere (hücre
dışı matris) bağlayan bir çapa görevi görür.
SGCA gen
mutasyonu, protein yapı bloğunun (amino asit) argininini, Arg77Cys veya R77C
olarak yazılan alfa-sarkoglikan proteininde 77. pozisyondaki amino asit sistein
ile değiştirir. Bilinen SGCA gen
mutasyonlarının geri kalanı, bireysel ailelere veya belirli
popülasyonlara özgüdür.
SGCA gen
mutasyonları sarkoglikan kompleksinin distrofin kompleksini oluşturmasını veya
bunstrofin kompleksine bağlanmasını ve stabilize olmasını önleyebilir. Bu
komplekslerle ilgili problemler kas liflerinin gücünü ve esnekliğini azaltarak
Kol-Kuşak Kas Distrofisine neden olur.
Belirti
ve Semptomlar
Bu kısımda Kol-Kuşak
Kas Distrofisi tip 2D hastalığının belirtileri listelenmiştir. Ancak çoğu
hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan
insanlar listelenen tüm semptomlara sahip olmayabilir.
İnsanların %30 -79 bu belirtilere sahiptir:
Aşil tendonu kontraktürü
(kısalması)
Buzağı kas psödohipertrofisi
Merdiven çıkma zorluğu
Yüksek serum kreatin kinaz (dolaşımda yüksek
kreatin fosfokinaz enzim düzeyi)
Sık düşme
Bu belirtilerin yanında skapular kanatlanma, hiperlordoz, sınırlı omuz hareketi, proksimal kas güçsüzlüğü, ayak yürüyüşü, vatka yürüyüşü gibi belirtiler de görülür. Nadir olarak torasik skolyoz da gözlenmiştir.
LGMD2D’nin klinik spektrumu 10-12 yaşlarında
ambulasyon (ayağa kalkma, yürüme) kaybına yol açan erken başlangıçlı ve hızlı
ilerleyen hastalar (a), ergenlikte yavaş
ilerleyen çocukluk başlangıçlı formları (b, c) ve yetişkinlikte başlayan
miyopatik değişikliklere sahip daha hafif formlar (d)
Kol-Kuşak Kas Distrofisinin prevalansını belirlemek zordur, çünkü özellikleri değişir ve
diğer kas bozukluklarınınki ile çakışır. Prevalans tahminleri 14.500’de 1
ile 123.000 kişide 1 arasında değişmektedir.
Kalıtım
Paterni
Kol-Kuşak Kas Distrofisi Tip 2D, otozomal resesif bir
paternde kalıtsaldır. Yani her bir
hücredeki genin her iki kopyasının mutasyonları vardır. Resesif genetik bozukluklarda hasta birey, her bir
ebeveynden aynı özellik için aynı anormal geni miras aldığında ortaya
çıkar. Bir kişi hastalık için bir normal gen ve bir gen alırsa, kişi hastalık
için bir taşıyıcı olacaktır ancak genellikle hastalığın belirtilerini göstermez. İki
taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip
olma riski her hamilelikte % 25’tir. Her hamilelikte ebeveyn gibi taşıyıcı
olan bir çocuk sahibi olma riski % 50’dir. Bir çocuğun her iki ebeveynden
de normal gen alma ve bu özellik için genetik olarak normal olma şansı %25’tir. Risk
erkekler ve kadınlar için eşittir.
Teşhis
Yöntemleri
LGMD (Kol-Kuşak Kas
Distrofisi) grubunda, hastaya ve ailesine doğru genetik tavsiye verilebilmesi
ve hastalıkların varlığından hastalık varlığına değişebilen komplikasyonların
yönetimi için uygun rehberlik sağlanabilmesi için kesin bir tanıya ulaşmak
önemlidir. Bu özellikle kardiyak veya solunumsal komplikasyon riski ile
ilgilidir. Bugün mevcut olan kesin test, geçmişte LGMD tanısı konan
bireylerin yeniden değerlendirilmesini ve daha kesin bir moleküler tanı
verilmesini mümkün kılabilir.
LGMD tanısı, kapsamlı bir
klinik değerlendirme, ayrıntılı bir hasta öyküsü, karakteristik semptomların
tanımlanması (örn. kas zayıflığı ve atrofinin spesifik dağılımı) ve cerrahi
olarak çıkarılması ve mikroskopik muayenesi (biyopsi) dahil olmak üzere çeşitli
özel testlere dayanarak konur. Bu özel testlere kasların sağlığını ve kasları
kontrol eden sinirleri değerlendiren bir test olan elektromiyografi, özel
kan testleri, belirli kas proteinlerinin varlığını ve sayısını değerlendiren
testler (immünohistokimya) ve moleküler genetik test örenk verilebilir.
Bir elektromiyografi
sırasında, bir iğne elektrodu deriden etkilenen bir kas içine
sokulur. Elektrot kasın elektriksel aktivitesini kaydeder. Bu kayıt,
bir kasın sinirlere ne kadar iyi tepki verdiğini gösterir ve kas zayıflığının
kasın kendisinden mi yoksa kasları kontrol eden sinirlerden mi kaynaklandığını
belirleyebilir. Elektromiyografi testi, belirli bir LGMD alt tipinin
teşhisine izin vermez, ancak alternatif teşhisleri dışlamak için yararlı
olabilir.
Kan testleri, kas hasar
gördüğünde anormal derecede yüksek seviyelerde bulunan bir enzim olan yüksek
kreatin kinaz (CK) seviyelerini ortaya çıkarabilir. Yüksek CK seviyeleri,
tüm LGMD vakalarında olmasa da bazılarında görülür. LGMD’nin otozomal
resesif formlarında CK düzeyleri, otozomal dominant formlardan çok daha
yüksektir. Yüksek CK seviyelerinin tespiti, kasın hasar gördüğünü veya
iltihaplandığını doğrulayabilir, ancak LGMD tanısını doğrulayamaz. Bununla
birlikte, hangi tip LGMD’nin diğerlerinden daha olası olduğunu belirtmek
yardımcı olabilir.
Moleküler genetik test,
spesifik bir genetik mutasyonu tanımlamak için deoksiribonükleik asitin (DNA)
incelenmesini içerir. Bu artık LGMD’de tanı için altın standarttır ve
diğer aile üyeleri için spesifik bir tanıya ve spesifik testlere izin verir.
LGMD vakıflarından oluşan bir
konsorsiyum, kas güçsüzlüğü olan hastalara ücretsiz genetik sekans
sunmak için http://LGMD-diagnosis.org adresinde teşhis programı oluşturdu. Kas
güçsüzlükleri için genetik bir açıklaması olmayan bireylerin, serbest genetik
sekanslama için uygun olup olmadıklarını belirlemek için alabilecekleri
çevrimiçi bir test sunmaktadır. Doktorlar, hastalarının uygun olup
olmadığını belirlemek için Jain Vakfı tarafından geliştirilen Otomatik LGMD
Teşhis Asistanını (ALDA) kullanarak hastaları adına da başvurabilirler.
Tedavi Yöntemleri
LGMD’nin herhangi bir formu
için herhangi bir tedavi yoktur. Tedavi, her bir kişide mevcut olan
spesifik semptomlara yöneliktir. Özel tedavi seçenekleri arasında kas
gücünü artırmak ve kontraktürleri (kısalmaları) önlemek için fiziksel ve
mesleki terapi, yürüme (ambulasyon) ve hareketliliğe yardımcı olmak için
çeşitli cihazların (örn. bastonlar, diş telleri, yürüyüşçüler, tekerlekli sandalyeler)
kullanılması ve skolyoz gibi iskelet anormalliklerini düzeltmek için
cerrahi gibi yöntemler yer almaktadır.
Genetik danışmanlık etkilenen
bireyler ve aileleri için yararlı olabilir. Diğer tedaviler semptomatik
(belirtilere yönelik) ve destekleyicidir. Hastalara ilgili hasta
organizasyonları ve kayıtları için iletişim bilgileri verilmelidir.
Hastalıkla
İlişkili Genler
SGCA geni (GKRY)
Hastalığın
Diğer İsimleri
LGMD
Tip 2D
LGMD2D
LGMDR3
Pelvofemoral Kas Distrofisi
Proksimal Kas Distrofisi
Alfa-sarkoglikan ile Kas Distrofisi Uzuv Kemeri
Alfa-sarkoglikan
ile ilgili LGMDR3
Alfa-sarkoglikan
eksikliğine bağlı LGMD
Alfa-sargoglikan
eksikliğine bağlı Ekstremite Kuşak Kas Distrofisi
Alfa-sarcoglycanopathy
Duchenne Benzeri Otozomal Resesif Kas Distrofisi, Tip 2
Alfa-mannosidoz
(α-mannosidoz), lizozomal α-D-mannosidaz enzimini kodlayan gendeki
mutasyonların neden olduğu otozomal resesif kalıtım ile seyrek görülen bir
lizozomal depo hastalığıdır. Zihinsel yetersizlik, işitme kaybı, ataksi,
iskelet anormallikleri ve anormal yüz özellikleri ile karakterizedir. Belirti
ve semptomlar hastalığın şiddetine göre değişiklik gösterebilir. Genellikle
hafif ila orta derecede zihinsel engel, işitme kaybı, zayıflamış bağışıklık
sistemi, ayırt edici yüz özellikleri ve serebellar bozukluklar (örn. , ataksi)
görülür.
Belirti ve Semptomlar
Alfa-mannosidozun
semptomları, ilerlemesi ve şiddeti, aynı mutasyonu paylaşan kardeşler dahil
olmak üzere bir kişiden diğerine büyük ölçüde değişiklik gösterir. Bazı
bireylerde doğumdan kısa bir süre sonra semptomlar gelişebilir. Bebeklik veya
erken çocukluk döneminde potansiyel olarak hayatı tehdit eden komplikasyonlar
gelişebilir. Diğer bireylerde ise genellikle 10 yaşından önce başlayan daha
ılımlı semptomlar gelişebilir. Bazı durumlarda ise bireyler yetişkinliğe kadar
teşhis edilemeyebilir. Genel olarak, etkilenen bireyler zihinsel engel, ayırt
edici yüz özellikleri ve iskelet anormalliklerine sahip olabilir. Semptomlar
zamanla yavaşça kötüleşebilir.
Semptomların
şiddetine göre 3 alt tipte sınıflandırılır:
Tip 1: Kas problemleri (miyopati) ile yavaş ilerlemenin olduğu on yaşından sonra gelişen semptomları içeren hafif bir formdur.
Tip 2: İskelet anormallikleri, miyopati ve yavaş ilerleme ile birlikte on yaşından önce semptomların görüldüğü ılımlı bir formdur. İskelet anormallikleri genellikle azalmış kemik yoğunluğu, kafatasının üstündeki kemiklerin kalınlaşması, omurgadaki kemiklerin deformasyonları, eğilmiş bacaklar, kemik ve eklemlerde deformasyonları içerir. En yaygın olan tiptir.
Tip 3: Progresif santral sinir sistemi tutulumu ve enfeksiyonundan dolayı gebelik kaybı veya erken ölüm olarak kendini gösteren şiddetli bir formdur.
Karakteristik yüz özellikleri arasında büyük bir kafa, belirgin alın, düşük saç çizgisi, yuvarlak kaşlar, büyük kulaklar, burnun düzleştirilmiş köprüsü, çıkıntılı çene, geniş aralıklı dişler, büyümüş diş etleri ve büyük dil sayılabilir.
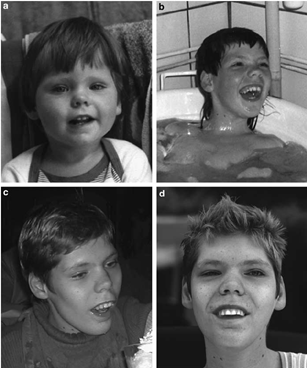
Şekil 1. Alfa-mannosidozda yüz özellikleri. A. 18 aylık ikizler. Genişlemiş baş, kısa boyun, yuvarlak kaşlar, yayılmış burun ve belirgin alın görülmektedir. B. Aynı ikizler 8 yaşında. Ellerin hafif kas atrofisi dikkat çeker. C. İşitme cihazı kullanan 27 yaşındaki alfa-mannosidoz hastası.
Diğer
belirtiler arasında şunlar olabilir:
Hareketleri
koordine etmede zorluk (ataksi)
Oturma
ve yürüme gibi motor becerilerinde gecikme
Konuşma
bozuklukları
İmmün
yetmezlik nedeniyle artan enfeksiyon riski
Karaciğer
ve dalağın genişlemesi (hepatosplenomegali)
Beyindeki
sıvı birikmesi (hidrosefali)
İşitme
kaybı
Göz
merceğinin bulanıklaşması (katarakt) ve yakını görememe gibi göz problemleri
Eklem
ağrısı ve iltihabı
Alfa-mannosidozu
olan bazı kişilerin depresyon, anksiyete veya halüsinasyonlar gibi zihinsel sorunları
vardır. Kalp ve böbrek problemleri de ortaya çıkabilir.
Prevalans
Alfa-mannosidozun
prevalansı hakkında çok az şey bilinmektedir. Avustralya’da yapılan bir
araştırmada, 500,000 kişide bir yaygınlık olduğu bildirilmiştir. Norveç’te yapılan
bir çalışmada 4.5 milyonluk bir nüfusta sadece altı kişide bu hastalığın
varlığı bildirilmiştir. Ayrıca Çekya’da 300,000 kişide bir kişi olarak
bildirilmiştir. Hastalık herhangi bir etnik gruba özgü değildir; dünyanın her
yerinden bireylerde tanımlanmıştır.
Alfa-mannosidozun
ayrıca dünya çapında yaklaşık 500,000 kişiden birinde meydana geldiği tahmin
edilmektedir.
Kalıtım Paterni
MAN2B1
genindeki mutasyonlar alfa-mannosidoza neden olur. Bu gen, alfa-mannosidaz
enzimini yapmak için talimatlar sağlar. Bu enzim lizozomlarda çalışır. Enzim
lizozomlar içinde, belirli proteinlere (glikoproteinler) bağlı oligosakkaritlerin
parçalanmasına yardımcı olur. Özellikle, alfa-mannosidaz enzimi mannoz adı
verilen şeker molekülü içeren oligosakkaritlerin parçalanmasına yardımcı olur.
MAN2B1 genindeki mutasyonlar, alfa-mannosidaz enziminin mannoz içeren oligosakkaritlerin parçalanmasındaki rolünü yerine getirme kabiliyetine müdahale eder. Bu oligosakkaritler lizozomlarda birikir ve hücrelerin arızalanmasına ve sonunda ölmesine neden olur.
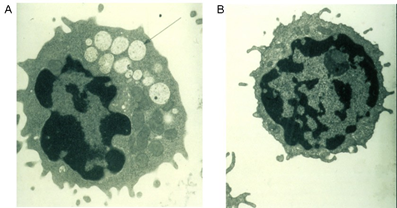
Şekil 2. Oligosakkarit birikimi sonucu koful oluşturan bir lenfosit ve normal lenfosit elektron mikrografisi. A. Alfa-mannosidoz hastasında koful oluşturan bir lenfosit. B. Normal olan bir lenfosit.
Dokular
ve organlar, oligosakkaritlerin anormal birikimi ve ortaya çıkan hücre ölümü
nedeniyle hasar görür ve alfa-mannosidozun karakteristik özelliklerine yol
açar.
Teşhis Yöntemleri Ve Tedaviler
Lizozomal
depo hastalığı olan alfa-mannosidozdaki ana klinik özellikler diğer lizozomal
depo hastalıkları ile örtüşebilir. Bununla birlikte, bu diğer lizozomal depo
hastalıkları ile ilişkili ayırt edici klinik özellikler, klinik
laboratuvarlarda biyokimyasal testlerin mevcudiyeti ve doğal geçmişlerinin
anlaşılması, bunların birbirinden ayırt edilmesine yardımcı olmaktadır.
Alfa-mannosidoz
ön tanısı konulan bir kişide hastalığın ve hastalık için gerekli kişisel
tedavilerin belirlenmesi için, aşağıdaki değerlendirmeler önerilir:
Hastanın
işitme kaybı, sinirlilik, depresyon; sosyal, ev, okul veya işle ilgili
aktivitelerde veya yürüme mesafesindeki değişiklik; ishal veya idrar tutamama,
kas ağrısı, eklem ağrısı, azalmış hareket aralığı ve kemik ağrısı gibi
şikayetlerinin tıbbi öyküsünün belirlenmesi gerekmektedir.
Otoskopi,
oftalmoskopi, karaciğer ve dalak boyutunun değerlendirilmesi, kalp ve
akciğerlerin oskültasyonu, yürüyüş dahil nörolojik durum ve eklem hareket
açıklığının ortopedik değerlendirmesini içeren fizik muayene yapılmalıdır. Çocuklarda
standart büyüme çizelgeleri kullanarak boy, ağırlık ve özellikle baş çevresinin
büyüklüğü ölçülüp büyüme değerlendirilmelidir.
İşitme
bozukluğu ve orta kulak enfeksiyonlarını tespit etmek için bir kulak burun
boğaz uzmanı tarafından muayene edilmelidir.
Kornea
opasiteleri, miyopi, hipermetropi ve şaşılık açısından oftalmolojik olarakdeğerlendirilmelidir.
Fonksiyonel
seviyeyi ve öğrenme kapasitesini belirlemek için nöropsikolojik testler
uygulanmalıdır.
Sistemik
lupus eritematozusu (SLE) hariç tutmak için klinik muayene ve immünolojik
testler (örn., Antinükleer antikorlar, anti-ds-DNA antikorları) ile kan
testleri yapılmalıdır.
Başın
düz radyografileri, dizler (ön-arka görünüm), omurga (yan görünüm) ve herhangi
bir semptomatik bölgenin iskelet değerlendirmesi yapılmalıdır.
Yaşlı
bireylerde osteopeni veya osteoporozu tespit etmek için kemik dansitometrisi
uygulanmalıdır.
Özellikle
hidrosefali belirtileri ve semptomları varsa (örneğin, baş ağrısı, artan
yürüyüş ataksisi, bulantı) ventriküllerin boyutunun ve serebellumun şeklinin ve
boyutunun değerlendirilmesi için beynin BT taraması yapılmalıdır.
Bir
klinik genetik uzmanı ve / veya genetik danışman ile konsültasyon.
Çok
semptomatik bir hastalığın karakteristik bulgularının tanımlanması üzerine
aşağıda açıklanan testlerin sonuçlarına dayanarak alfa-mannosidoz tanısı konulmasına
yardımcı olunabilir.
İdrarda
oligosakkaritler: İdrardaki mannoz açısından zengin oligosakkarit
konsantrasyonlarını ölçmek için bir ön araştırma yapılabilir. Mannoz zengini
oligosakkaritlerin yüksek idrar atılımı düşündürücüdür, ancak hastalığın tanısı
için yeterli değildir.
Alfa-mannosidaz
aktivitesi: Tanı, bir florometrik analiz yoluyla lökositlerde veya diğer
çekirdekli hücrelerdeki alfa-mannosidaz aktivitesinin ölçülmesi ile doğrulanır.
Bu test, genetik testle birlikte en güvenilir tanı yöntemidir.
Genetik
test: Hastalığa neden olan mutasyonun, periferik kan örneği kullanılarak polimeraz
zincir reaksiyonu (PCR) amplifikasyonu ile tanımlanması güvenilir bir tanı
yöntemidir.
Uygulanan Tedaviler
Konjenital
alfa-mannosidoz için bir tedavi yoktur ve genel olarak, ortaya çıkan
komplikasyonları önlemek amacıyla hastalığın yönetimi ele alınır. Aşılar,
antibiyotikler, işitme cihazları, gözlükler, ortopedik ve diğer yardımcı
cihazlar, eğitim müdahaleleri ve konuşma terapisi gibi bireysel semptomlara
yönelik tedaviler gerektiği şekilde önerilir. Sağlığı ve tedaviye yanıtı
izlemek için düzenli takip önerilmektedir.
Araştırılmakta Olan Tedaviler
Kemik iliği nakli
Alfa-mannosidoz
tedavisinde kemik iliği nakli denenmiştir. Kemik iliği nakli ile erken tanı ve
hızlı tedavi, bilişsel gerilemeyi önleme ve semptomları iyileştirme şansını
arttırır. Bununla birlikte, bu potansiyel tedavinin uzun vadeli güvenliğini ve
etkinliğini belirlemek için daha fazla araştırmaya ihtiyaç vardır. Prosedürü
ciddi komplikasyon riski taşır. Kemik iliği naklinden sonra, normal gelişim
sağlanamamasına rağmen, etkilenen bireyler gelişimsel ilerleme kaydetmiştir. En
iyi donör ailesel HLA-özdeş olandır, ancak çoğu zaman bu tip donör
tanımlanamaz, bu durumda enzim replasman tedavisi (ERT) en iyi seçenek olarak
belirlenebilir.
Gen Terapisi
Gen
terapisi ayrıca bazı lizozomal depo bozuklukları için olası bir tedavi olarak
araştırılmaktadır. Aktif enzim üretebilen normal genin kalıcı transferi göz
önüne alındığında, bu tedavi şeklinin teorik olarak bir tedaviye yol açması
muhtemeldir. Bununla birlikte, şu anda, gen terapisi başarılı olmadan önce
çözülmesi gereken birçok teknik zorluk vardır.
Allan – Herndon –
Dudley sendromu (AHDS) ciddi derecede zihinsel gelişim, dizartri, atetoid
hareketler, kas hipoplazisi ve spastik parapleji ile karakterize X’e bağlı bir
durumdur. Sadece erkeklerde meydana
gelen bu durum, gelişimi doğumdan önce bozar. Hastalık, etkilenen erkeklerde
konuşmayı ve iletişim yeteneğini kısıtlamış olsa da, diğer insanlarla iletişimde
eğleniyor gibi görünüyorlar.
Allan-Herndon-Dudley
sendromlu çocukların çoğunda zayıf kas tonusu (hipotoni) ve birçok kasın az
gelişmesi (kas hipoplazisi) durumu vardır. Yaşlandıkça, genellikle belirli
eklemlerin hareketini kısıtlayan kontraktür denilen eklem bozuklukları
geliştirir. Anormal kas sertliği (spastisite), kas zayıflığı ve kolların ve
bacakların istemsiz hareketleri de hareketliliği sınırlar. Sonuç olarak,
Allan-Herndon-Dudley sendromlu birçok insan bağımsız olarak yürüyemez ve yetişkinlikte
tekerlekli sandalye kullanımı mevcuttur.
Klinik Tanım
Hastalık
spastisiteye (kontraktürler, Babinski işareti ve klonus) ilerleyen ve
genellikle yaşamın erken döneminde saptanabilen konjenital hipotoni (doğumda
veya yaşamın ilk haftalarında / aylarında görülür) olarak kendini gösterir.
Hiperrefleksi, daha sonra yaşamda ortaya çıkar. Etkilenen erkekler de bebeklik
ve erken çocukluk döneminde, motor dönüm noktaları gecikmesi ve baş ve
desteklemede zorluk olarak ortaya çıkan kas hipoplazisi ve genel kas zayıflığı
ile kendini gösterir. Hastaların% 100’ünde hipotoni ve ciddi zihinsel eksiklik
vardır. Şiddetli psikomotor gecikme en başından itibaren mevcuttur (motor ve
dil dönüm noktalarının gecikmesi) ve özerkliğe asla ulaşılamaz. Yüz, zaman
içinde gelişen ayırt edici özelliklere sahiptir: açık ağız, çadırlı üst dudak, pitoz(sarkma)
, kulakların anormal katlanması, burun ve kulakların yumuşak dokusunun
kalınlaşması ve kalkık kulak memeleri. Uzun, ince eğik ayaklar da tipiktir.
Oküler belirtiler (yani döner nistagmus ve ayrık göz hareketleri) nadirdir.
Nöbetler ve kilo alım problemleri bazı hastalarda bildirilmiştir. Hipotoni ve
kas hipoplazisinin bir sonucu olarak düşünülen , pektus ekskavatum ve skolyoz
bulunabilir.
Epidemeyoloji
Bugüne kadar
literatürde 320 hastalıktan etkilenen bireye sahip en az 132 aile
bildirilmiştir. Yaygınlık bilinmemekle birlikte, bir çalışma, etiyolojisi
bilinmeyen zihinsel özürü olan erkeklerin% 1.4’ünde AHDS’yi tanımlamıştır.
Sadece erkekler etkilenir.
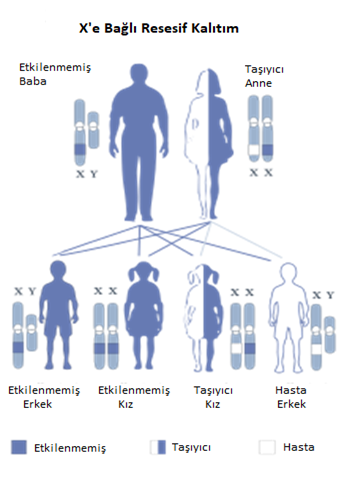
Kalıtım Kalıbı
Bu durum X’e bağlı resesif, kalıtsaldır. Hastalığa neden
olan mutasyona uğramış gen , iki cinsiyet kromozomundan biri olan X kromozomu
üzerinde bulunuyorsa, durum X’e bağlı olarak kabul edilir. Sadece bir X
kromozomu olan erkeklerde , her hücrede genin değiştirilmiş bir kopyasının
bulunması, hastalığa neden olmak için yeterlidir. İki X kromozomu olan
kadınlarda, bozukluğa neden olmak için genin her iki kopyasında da mutasyon
bulunmalıdır. Erkekler X’e bağlı resesif bozukluklardan kadınlardan daha sık
etkilenir. X’e bağlı kalıtımın bir özelliği, babaların X’e bağlı özellikleri,
oğullarına geçirememesidir.
Teşhis Yöntemleri
Tanı, klinik
bulgulara ve diğer tiroid hormonu serum düzeylerinin varlığına dayanır:
erkeklerde anormal derecede yüksek 3,3 ‘, 5’-triiyodotironin (T3), düşük ila
normal serbest tetraiyodotironin (T4) seviyeleri ve normal ila hafif yüksek
tiroid uyarıcı hormon (TSH) seviyeleri vardır. SLC16A2 genindeki mutasyonları
gösteren moleküler genetik test tanıyı doğrular.
Yönetim ve Tedavi
Şu anda, AHDS için
herhangi bir tedavi mevcut değildir ve yönetim destekleyici önlemlerden oluşur.
Fiziksel, mesleki ve konuşma terapisi faydalı olabilir. Distoni,
antikolinerjikler, L-DOPA, karbamazepin veya lioresol gibi bazı ilaçlarla
tedavi edilebilir. Nöbetler mevcut
olduğunda, standart antiepileptik ilaçlarla kontrol edilebilir. Hipotiroidizm
tedavisi yararlı görünmemektedir.
Hastalıkla İlgili Genler
AHDS, tiroid hormonu
T3 için spesifik bir taşıyıcı olan monokarboksilat taşıyıcı 8’i (MCT8) kodlayan
SLC16A2 genindeki (Xq13.2) mutasyonlardan
kaynaklanır . Nörolojik sorunlar, tiroid hormonu T3’ün bazı nöronal hücrelere
taşınamamasından kaynaklanabilir.
Prognoz
Birkaç hasta
60’larında hayatta kalmasına rağmen, çoğu hasta bağımsız olarak oturamadığı,
ayakta duramadığı veya yürüyemediği için genel yaşam beklentisi tehlikeye
giriyor ve yaşam kalitesi ciddi şekilde etkileniyor.
Oküler albinizm tip I (OA1) veya X’e
bağlı oküler albinizm, oküler albinizmin en yaygın şeklidir. İlk kez 1909’da
Nettleship tarafından tanımlanmıştır. Oküler albinizm, X’e bağlı resesif bir
genetik hastalık olarak kalıtsaldır. X’e bağlı olduğu için hastalık büyük
çoğunlukla erkeklerde görülür ve görme bozuklukları ile karakterize genetik bir
hastalıktır. Görme bozuklukları doğumda mevcuttur yani konjenitaldir ve zamanla
daha şiddetli hale gelmez. Oküler albinizm, öncelikle gözdeki pigment üretimini
etkiler. Sinirlerde retinadan beyne giden anormal bağlantılar, gözlerin
birlikte hareket etmesini, izlemesini önler ve derinlik algısını azaltır.
Gözler istemsiz bir şekilde ileri geri hareket edebilir.
[1,3]
Belirti ve Semptomlar,
Oküler albinizm öncelikle gözdeki
pigment üretimini etkiler. Oküler albinizmde gözlerin ileri geri hareketi
(nistagmus), görme bozukluğu, bazı kişilerde yarı saydamlıkla birlikte gelişen iris pigmentinin azalması (hipopigmentasyon),
retina pigmentinin azalması, albinotik fundus, maküler hipoplazi, fovea
gelişiminin olmaması (foveal hipoplazi) dahil olmak üzere çeşitli görme
sorunları ortaya çıkabilir. Hastalarda derinlik algısında azalma meydana
gelebilir. Gözlerin birlikte hareketi ve izlemesi azalabilir. Çapraz gözler
(şaşılık) ve ışığa duyarlılık (fotofobi) de yaygındır. X’e bağlı konjenital nistagmus 6, oküler
albinizm ile birlikte gelişebilen bir allelik hastalıktır. Etkilenen bireyler,
görüş netliğini artırmak için genellikle başlarını çevirir veya sallar. Görme
bozuklukları doğumda mevcuttur ve zamanla daha şiddetli hale gelmez. Etkilenen
bireyler genellikle normal cilt ve saç pigmentasyonuna sahiptir.
İlerleyen zamanlarda yapılan çalışmalar
ve araştırmalar ile oküler albinizmin yalnızca gözleri değil melanozomlar
(melanin içeren organel) üzerinde de olumsuz bir durum oluşturabileceği
gösterilmiştir. X’e bağlı oküler albinizmi olan Kafkaslar ve siyah tenliler ile
yapılan histopatolojik bazı çalışmalar ile, hasta erkeklerin ve heterozigot
dişilerin, epidermis melanositlerinde ve keratinositlerinde dev melanozomların
varlığı belirlenmiştir (O’Donnell ve ark., 1976; O’Donnell ve ark., 1978;
Garner ve Jay, 1980). Etkilenen erkekler bazen ciltlerinde hafif bir hipopigmentasyon
gösterir ve sıklıkla vücutlarında konjenital hipopigmente maküller (leke,
lezyon) ve bölgeler vardır.
X’e bağlı kalıtılar bir hastalık olduğundan, hastalığın sonuçları,
semptomları heterozigot veya homozigot olma durumuna göre ve kadın veya erkek
olma durumuna göre değişiklik göstermektedir.
Etkilenen
erkekler görüş keskinliğinde azalma, yarı saydam irisler, konjenital nistagmus,
fotofobi, foveal refleksleri olmayan fundusun hipopigmentasyonu ve yüksek
şaşılık insidansına sahiptir. Renk körlüğü etkilenen bazı erkeklerde
belirtilmiş olsa da (Forsius and Erkisson,1964), çoğunlukla renk görmede bir
sorun meydana gelmemektedir.
X’e bağlı oküler albinizm için heterozigot olan dişiler yani taşıyıcılar, normal görsel fonksiyonlarına rağmen, genellikle funduslarında (göz yuvarlağının arkasındaki alan) karakteristik ve sıklıkla çarpıcı değişikliklere sahiptir. Bu değişiklikler normal veya hiperpigmente retinanın bölgeleri arasına serpiştirilmiş periyotta hipopigmentasyon alanlarından oluşurken, arka kutuplar genellikle pigment epitelinde bir beneklenme gösterir. Normal fundus durumları da heterozigotlukta gösterilmiştir. Aynı zamanda heterozigot dişilerin irisleri sıklıkla yarı saydamlık göstermektedir.
(Şekil 1) [3]
X’e bağlı resesif oküler albinizmde, heterozigot durumda meydana gelen yarı saydam iris gösterilmiştir.
(Şekil 2) [3]
Heterozigot bir bireyin epidermisinin bazal laminası elektron mikroskopu gösterimiştir. Makromelanozom ok ile işaret edilmiştir (x 5.400).
(Şekil 3) [5]
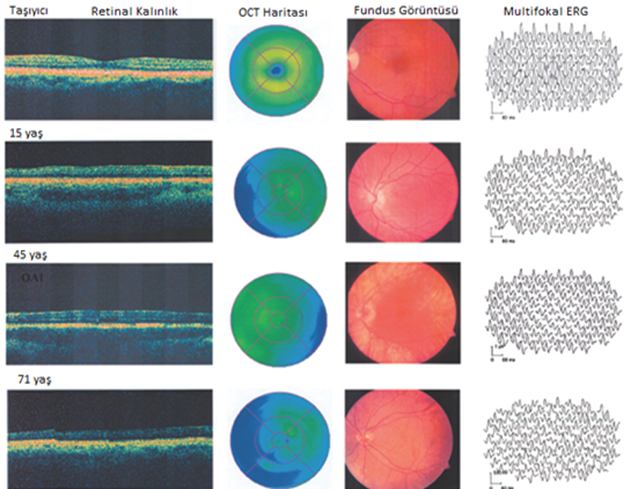
Üç farklı yaştaki erkek hastanın retinal
kalınlığı, OCT haritası (optik koherens tomografi), fundus görüntüsü ve
multifokal elektroretinografi (ERG) sonuçları verilmiştir. Bir tane de heterozigot
taşıyıcı sonuçları da karşılaştırma yapılması açısından eklenmiştir.
[1, 2, 3, 5]
Genetik
Oküler albinizm, X’e bağlı resesif bir genetik hastalık
olarak kalıtsaldır. Hastalığın insidansı 20.000 erkek çocuk doğumunda 1’dir.
404 amino asitlik bir proteini kodlayan,
X kromozomu üzerindeki (Xp22) G
proteinine bağlı reseptör 143 (GPR143) genindeki mutasyonlar, oküler albinizm
ile ilişkilendirilirler. Bu protein,
göz ve melanositlerin retinal pigment epitelinde (RPE) eksprese edilir. GPR143,
melanin pigment oluşumunun düzenlenmesinde rol oynayan premelanozomal protein
MART1 ile etkileşime girer. MART1, GPR143 için bir şaperon proteini olarak
işlev görebilir. GPR143’teki mutasyonlar, anormal fibril oluşumuna eşlik eden
genişlemiş anormal premelanozomlara neden olur. Premelanozom, pigment
hücresindeki melanin pigment üretiminin hücre içi konumudur. Premelanozomlarda melanin
pigment sentezinde de bir azalma vardır. Derideki melanozomlarında bozukluklar
da mevcuttur, ancak cilt ve saç pigmentinin miktarını azalttığı
görülmemektedir.
GPR143’ün, melanin pigment yolunda bir
ara metabolit olan L-DOPA (L-3,4-dihidroksifenilalanin) için bir reseptör
olduğu düşünülür ve retinada hücre içi sinyalleşmeye katılıyor olabilir. OA1
ile ilişkili mutasyonların çoğu eksik anlatım ile ilgili (missense)
mutasyonlardır, fakat anlamsız (nonsense), çerçeve kayması ve splice bölgesi
mutasyonları
(splice site mutations) da
rapor edilmiştir. Bir veya daha fazla ekson dahil olmak üzere birkaç büyük
delesyon da bildirilmiştir.
Bazı durumlarda SHROOM2 gibi flanking
genler de dahil edilebilir, ancak bu ilave genlerin pigmentasyondaki
değişikliklerle ilişkili olmadığı, ancak oküler albinizm tip 1 sendromunda yer
alabileceği gösterilmiştir.
Dişilerin iki X kromozomu vardır, ancak X kromozomlarından biri “kapatılır” ve bu kromozom üzerindeki genlerin çoğu inaktive edilir. X kromozomlarından birinde mevcut bir hastalık geni olan dişiler bu hastalığın taşıyıcılarıdır. Taşıyıcı dişiler genellikle bozukluğun belirtilerini göstermez, çünkü X kromozomunun inaktivasyonu rastgeledir ve genellikle gözdeki hücrelerin yarısında normal X kromozomu aktifleşir ve normal görme ile sonuçlanır.
(Şekil 4) [6]
X’e bağlı resesif bir hastalık
olduğundan erkekler çoğunlukla hasta olurken kadınlar taşıyıcı olabilirler.
Baba hasta, anne hasta ise ise kız çocuk da %100 oranda hasta olur. Kız çocuğa
heterozigot bir şekilde geldi ise taşıyıcı olur. Anne taşıyıcı ise erkek çocuklar
%50 hasta, hasta ise %100 hasta olur.
Bazı GPR143 mutasyonlarının erkeklerde
X’e bağlı konjenital nistagmus ile sonuçlandığına dair kanıtlar vardır.
Tanı
Oküler albinizm tanısı
karakteristik göz bulgularına dayanmaktadır.
-Erkekler için:
Oftalmolojik testler
yapılır. İnfantil nistagmus, irisin
hipopigmentasyonu, oküler fundusun hipopigmentasyonu, foveal hipoplazi.
azaltılmış görme keskinliği, anormal optik yol görüntüleri açısından
incelemeler yapılır.
İnfatil nistagmus, genellikle yaşamın
ilk üç ayında gelişir ve yaşamın ilerleyen döneminde semptom bir düşüş gösterse
de nadiren tamamen düzelir. Genellikle yakın mesafeden görüş, uzak mesafeden
görüşe göre daha iyidir. İris pigment epitelinin (IPE) hipopigmentasyonu, iris
transillüminasyonuna sebep olur. İris pigment epiteli, irisin arka tabakasındadır.
Oküler fundusun hipopigmentasyonu, retinal pigment epitelinde (RPE) azalmış
pigment konsantrasyonundan kaynaklanır. Foveal hipoplazi, foveal çukurun (umbo)
ve halka şeklindeki foveal refleksin azalması veya yokluğu ile karakterizedir.
XLOA ilerleyici olmayan bir hastalıktır ve görme keskinliği tipik olarak
gençliğin ortasına veya biraz daha sonuna kadar yavaşça ilerler ve daha sonra
yaşam boyunca stabil kalır. Çeşitli albinizm formlarında MR görüntülemesi,
optik kiazmanın boyut ve konfigürasyonunda normal kontrollere göre farklılıklar
bulmuştur.
Dermatolojik
olarak, saçta ve deride hipopigmentasyo görülür. Deri renk spektrumu geniştir
ve diğer albinizm formlarının tüm spektrumlarını aşar.
-Heterozigot dişiler için:
Genel etnik ve ırka özgü deri ve adneksiyal
pigmentasyona bağlı olarak, heterozigot dişiler iris transillüminasyonunu ve
vasküler arcade dışında daha dramatik hale gelen retina pigment epitelinin
kalın lekeli hipo ve hiperpigmentasyonu gösterebilir. Bazı taşıyıcılar, izole
edilmiş hipopigmente cilt lekelerine sahiptir. Nadiren, infantil nistagmus,
foveal hipoplazi, azalmış görme keskinliği ve oküler yapıların dağınık
hipopigmentasyonunu gösteren heterozigot dişiler belirtilmiştir.
-Hastalığın moleküler tanısı:
Hastalığın
moleküler test ile tanısının konulması ve kesin bir şekilde kanıtlanması mümkün
olabilmektedir.
GPR143 geni için moleküler genetik test,
etkilenen erkeklerin yaklaşık %90’ındaki mutasyonları tespit eder ve tanıyı
doğrulamak için kullanılabilir. XLOA tanısı, GPR143’te patojenik
bir varyant tanımlaması ile bir probanda konulur. Moleküler test mevcut
değilse, cilt biyopsisinde makromelanozomların bulunması da tanıyı koyacaktır.
Moleküler test yaklaşımları, tek gen
testini ve bir multigen panelinin kullanımını içermektedir. Tek gen testinde GPR143’ün
önce sekans analizi yapılır, ardından patojenik bir değişken bulunmazsa gen
hedefli delesyon/dublikasyon analizi yapılır. Multigen panelde ise GPR143 ve
diğer ilgili genleri içeren çokgenli bir panel uygulanır. Panele dahil edilen
genler ve her bir gen için kullanılan testlerin tanısal duyarlılığı laboratuara
göre değişir ve zaman içinde değişmesi muhtemeldir. Panelde kullanılan
yöntemler sekans analizi, delesyon/dublikasyon analizi ve/veya sekanslama
tabanlı olmayan diğer testleri içerebilir.
Tedavi
Oküler
albinizm tanısı konulan bireyler hastalığın gidişatını, kapsamını belirlemek
için düzenli olarak muayeneye devam etmektedirler.
Gözlük veya kontakt lensler görüşü büyük ölçüde
arttırmaktadır. Güneş gözlüğü kullanımı veya göze siper olucak kenarı olan
şapkalar (snapback şapka gibi) güneş hassasiyetinin azaltılmasına yardımcı
olabilir. Gözde ışığın kırılması ile ilgili problemler, erkenden uygun gözlük
kullanımına başlanmasıyla düzeltilebilir. Nistagmus sebepli anormal baş
duruşunu düzeltmek için prizmatik gözlük önerilmektedir. Şaşılık için cerrahi
operasyon şart değildir ancak estetik bakımdan istenirse yapılabilmektedir.
Yaşa uygun güneş koruyucu losyonlar ve giysiler için
dermatolojik danışma verilmektedir.
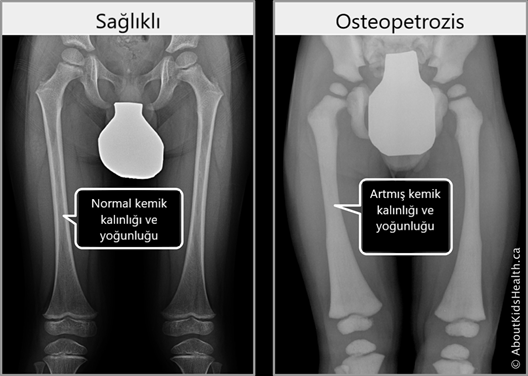
Osteopetrozis,
kemiklerin anormal şekilde yoğunlaşması ve kırılmaya yatkın olmasına neden olan
nadir, kalıtsal bir iskelet hastalığıdır. Semptomlar ve şiddetleri çok değişken
olabilir. Yenidoğanda ortaya çıkan, hayati tehlikesi olan belirtileri (kemik
iliği yetersizliği vb.) olabildiği gibi, tesadüfi olarak röntgende bulunarak da
teşhis konabilir. Semptomların şiddetine ve ortaya çıkma yaşına bağlı olarak
bireyde kemik kırıkları, kısa boy, kompresif nöropati (sinirlerde baskı),
tetanik kasılmaya eşlik eden hipokalsemi ve yaşamı tehdit eden pansitopeni (kan
hücrelerinin eksikliği) görülebilir. Nadir vakalarda nörolojik bozulma ya da
diğer vücut sistemlerinde de sorunlar görülebilir. Osteopetrozisin otozomal
dominant (baskın), otozomal resesif (çekinik) veya X’e bağlı olarak kalıtılan
tipleri vardır. Otozomal Dominant Osteopetrozis (Albers Schöngen Hastalığı),
genelde hastalığın en hafif tipidir. Etkilenen bireylerin bazılarında hiç
belirti görülmez. Belirti gösteren hastalarda ise kemik kırıkları, omurga
eğriliği (skolyoz) ya da diğer omurga anomalileri, kalçada eklem iltihabı
(artrit) ve kemik iltihabı (osteomiyelit) bulunabilir. Bu problemler genelde
çocukluk çağının sonlarında ya da ergenlikte belli olur. Tedavi yaklaşımları
semptomlara ve belirtilerin şiddetine göre değişiklik gösterir.
Belirti ve
Semptomlar
Belirtiler
kişiden kişiye birçok farklılık gösterebilir. Başlıca belirtiler şunlardır:
Kemiklerde şekil
anomalileri
Kemiği besleyen
damarlarda kan akımının azalması sonucu doku ölümü (avasküler nekroz)
Kemik ağrıları
Yüz felci (Bell
paralizi)
Eklem çıkıkları
Kafatasının
normalden büyük olması (makrosefali)
Görülme Sıklığı
Albers Schöngen Hastalığı, osteopetrozis tipleri arasında en sık görülenidir. 20,000 kişiden 1’ini etkiler. Otozomal dominant kalıtım gösterdiği için bireyde genin bir anormal kopyasının bulunması hastalığın oluşması için yeterlidir. Genelde mutasyonlu gen, hastalığa sahip ebeveynden aktarılır.
En az 9
gende meydana gelen mutasyonların, farklı tiplerdeki osteopetrozise neden
olabildiği bilinmektedir. CLCN7 genindeki
mutasyonlar, Otozomal Dominant Osteopetrozis vakalarının yaklaşık %75’inden
sorumludur. Diğer genlerdeki mutasyonlar daha az yaygındır. Osteopetrozis
vakalarının %30’unda hastalığın nedeni bilinmemektedir.
Osteopetrozisle
ilişkili genler, osteoklast denen özelleşmiş hücrelerin yapımından,
gelişiminden ve fonksiyonunun düzenlenmesinden sorumludurlar. Osteoklast
hücreleri, kemik yapım-yıkım döngüsünde (remodelasyon) kemik dokusunun
yıkımından sorumludur. Bu, eski kemik dokusunun yıkılıp onun yerine yeni kemik
oluşumunun gerçekleştiği normal bir döngüdür. Kemik remodelasyonu, kemiğin
sağlıklı ve güçlü kalmasını sağlamak amacıyla çok sıkı bir şekilde kontrol
edilir. Osteopetrozise neden olan genlerdeki mutasyonlar, osteoklast
hücrelerinin kaybına veya anormal hücrelerin oluşumuna yol açar. Düzgün işlev
göremeyen osteoklastlar, yeni kemik oluşurken eski kemiğin yıkımını sağlayamaz.
Sonuç olarak kemikler yoğun/ kalın bir hal alır. Ayrıca yapıları da bozuk olan
kemikler kırılmaya yatkın olurlar.
Teşhis Yöntemleri
Tanı, klinik bulgulara ve çoğu zaman iskeletin radyografik görüntülenmesine dayanır. Hastalığın kesin tanısı için ise genetik testler uygulanabilmektedir.
Alagille
sendromu, genellikle 5 ana klinik anormallik ile birlikte kolestaz, kalp
hastalığı, iskelet anormallikleri, oküler anormallikler ve karakteristik bir
yüz fenotipi ile tanımlanmış otozomal dominant bir hastalıktır. Kolestaz, safra
kanallarındaki bazı rahatsızlıkların doğrudan bir sonucudur. Hastaların
yaklaşık% 39’unda böbrek rahatsızlığı görülür, özellikle renal displazi vardır.
Ek semptomlar arasında kalp üfürümleri, konjenital kalp kusurları, vertebral
(sırt kemiği) farklılıkları, normalde gözdeki korneayı (arka embriyotokson)
hizalayan halkanın kalınlaşması ve ayırt edici yüz özellikleri bulunur.
Alagille sendromlu çoğu insanda JAG1 geninin bir kopyasında mutasyonlar vardır.
Hastaların küçük bir yüzdesi (yüzde 1’den az) NOTCH2 geninde mutasyonlara
sahiptir. Bu mutasyonlar otozomal dominant özellikler olarak miras alınır,
ancak vakaların yaklaşık yarısında mutasyon bireyde yeni bir değişiklik
(“de novo”) olarak ortaya çıkar ve bir ebeveynden miras alınmaz.
ALGS’nin şu anki tahmini insidansı cinsiyet açısından hiçbir fark olmaksızın 1:
30.000 ile 1: 45.000 arasındadır.
Hastalığın Semptonları
Alagille
sendromunun semptomları ve şiddeti, aynı ailenin üyeleri arasında bile bir
kişiden diğerine büyük ölçüde değişebilir. Bazı bireyler, neredeyse fark
edilmeyecek hafif bir bozukluğa sahip olabilir; diğer bireylerde potansiyel
olarak hayatı tehdit eden komplikasyonlara neden olabilecek ciddi bir hastalık
şekli olabilir. Etkilenen bireylerin aşağıda tartışılan semptomların hepsine
sahip olmayabileceğini belirtmek önemlidir. Etkilenen bireyler, özel vakaları,
ilişkili semptomları ve genel prognozu hakkında doktorları ve tıbbi ekipleriyle
konuşmalıdır. Alagille sendromu, karaciğer, kalp, gözler, iskelet, böbrekler ve
vücudun diğer organ sistemlerindeki anormalliklerle ilişkili olabilir. Alagille
sendromunun ana bulgusu, yaşamın ilk üç ayında sıklıkla ortaya çıkan karaciğer
hastalığıdır. Bununla birlikte, hafif karaciğer tutulumu olan bireylerde, daha
sonraki yaşamda tanı konulamayabilir. Alagille sendromunda karaciğer hastalığı,
varsa, sarılık veya hafif kolestazdan şiddetli, potansiyel olarak karaciğer
yetmezliğine neden olabilecek ciddi, ilerleyici karaciğer hastalığına kadar
değişebilir. Safra kanallarının sayısının azalması nedeniyle, Alagille
sendromlu bireyler genellikle yaşamın ilk dört ayında sarılık ve kolestaz
geliştirebilir. Kolestaz, karaciğerden safra akışının azalması veya engellenmesi
anlamına gelir. Kolestaz, cildin ve gözlerin beyazlarının sararmasına
(sarılık), cildin yüzeyinin hemen altında yoğun, soluk renkli dışkılar, koyu
renkli idrar, yağlı yaralar veya şişliklere (ksantomlar) neden olabilir ve
anormal genişlemiş karaciğer (hepatomegali) ve / veya genişlemiş dalak
(splenomegali). Vücut yağları ve yağda çözünen vitaminleri (A, D, E ve K
vitaminleri) düzgün bir şekilde ememediği için, etkilenen çocuklar da büyüme
eksiklikleri ve gelişmede yetersizlik yaşayabilir. Yaşamsal besin maddelerinin
malabsorpsiyonu ayrıca raşitizm, yumuşatılmış, zayıflamış kemikler (D vitamini
eksikliği), görme sorunları (A vitamini eksikliği), zayıf koordinasyon ve
gelişimsel gecikmelere (E vitamini eksikliği) ve kan pıhtılaşma sorunlarına (K
vitamini eksikliği) yol açabilir.
Genetik Etkenler
Alagille
sendromuna, iki genden biri olan JAG1 geni veya NOTCH2 geni mutasyonlarından
kaynaklanır. JAG1 geninin mutasyonları, vakaların yüzde 88’inden fazlasında
tanımlanmıştır. NOTCH2 genindeki mutasyonlar vakaların yüzde 1’inden azını
oluşturmaktadır. Bu mutasyonlar otozomal dominant özellikler olarak kalıtılır.
Bazı durumlarda, mutasyonlar kendiliğinden genetik değişiklik (yani yeni
mutasyon) nedeniyle rastgele oluşur.
Araştırmacılar,
Alagille sendromu vakalarının çoğunun, kromozom 20’nin (20p12) kısa kolunda (9)
bulunan JAG1 geninin mutasyonları nedeniyle ortaya çıktığını belirlediler.
Alagille sendromu vakalarının yaklaşık yüzde 6-7’sinde, bireylerin JAG1 geninin
tamamen silinmesi veya kaybı vardır. Bu bireyler, silinmenin ne kadar büyük
olduğuna ve kromozom 20 üzerindeki diğer genlerin ne olduğuna bağlı olarak daha
ciddi bir Alagille sendromu formuna sahip olabilir. Araştırmacılar, NOTCH2
geninin kromozom 1’in (1p13-p11) kısa kolunda bulunduğunu belirlediler.
Hastalığın
Görülme Sıklığı
Alagille
sendromu erkekleri ve kadınları eşit sayıda etkiler. Alagille sendromu
insidansının genel popülasyondaki 30.000-45.000 kişide yaklaşık 1 olduğu tahmin
edilmektedir. Bazı Alagille sendromu vakaları teşhis edilmemiş veya yanlış
teşhis edilmiş olabilir, bu da genel popülasyonda Alagille sendromunun gerçek
sıklığını belirlemeyi zorlaştırır.
Hastalığın Standart
Tedavileri
Alagille
sendromunun tedavisi, her bir kişide belirgin olan spesifik semptomlara
yöneliktir. Tedavi, bir uzman ekibinin koordineli çabalarını gerektirebilir.
Çocuk doktorları, gastroenterologlar, kardiyologlar, göz doktorları ve diğer
sağlık uzmanlarının, çocuğun tedavisini sistematik ve kapsamlı bir şekilde
planlaması gerekebilir. Alagille sendromlu bireylerin kalp, karın ve böbrek
anomalilerini taramak için karın ultrasonu ve bir tarama gözü (oftalmoloji)
muayenesi için temel bir ekokardiyogram (kalbin ultrasonu) olması gerekir.
Spesifik semptomlar, anestezi veya sedasyona gerek kalmadan çalışma boyunca oturabilecek
kadar yaşlı çocuklar için başın kan damarlarının (MRI / MRA) bir tarama
görüntüleme çalışması önerilir. Malabsorbsiyonu olan bireyler için vitamin ve
besinlerle tamamlayıcı tedavi şarttır. Bu tedavi A, D, E ve K vitaminlerini
geri yüklemeyi içerebilir. Küçük çocuklara orta zincirli trigliseritlerle
formül verilebilir, çünkü bu yağ formu kolestaz olan Alagille sendromlu kişiler
tarafından daha iyi emilir. Bazı durumlarda, etkilenen çocukların burundan
mideye giden bir tüpten (nazogastrik tüp) veya karın duvarındaki ve midedeki
küçük bir insizyonla (gastrostomi tüpü) doğrudan mideye yerleştirilen bir tüp
yoluyla ekstra kalori alması gerekebilir.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
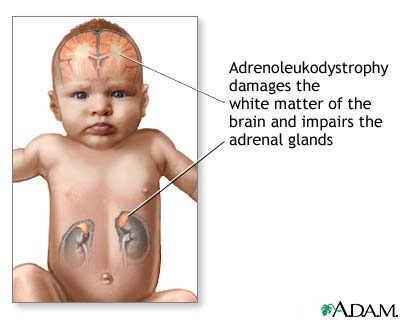
Yenidoğan adrenolökodistrofi (NALD), adrenal bezler(böbrek üstü bezleri) ve beynin ak maddesi dahil olmak üzere çoklu organları etkileyen kalıtsal bir hastalıktır.
Figür 1:NALD,beyin ak maddesine
zarar verir ve böbrek üstü bezlerinin işleyişini bozar.
NALD; Peroksizomların
biyogenezindeki ve/veya işleyişindeki
kusurlardan kaynaklanan ve peroksizom biyogenez bozuklukları(PBD) olarak
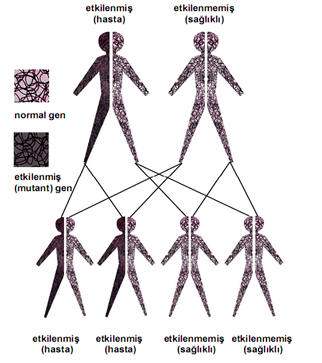
adlandırılan bir bozukluk ailesinin üyesi olan otozomal resesif bir bozukluktur.
Peroksizomlar, neredeyse tüm ökaryotik hücrelerde bulunan lizozomlara benzer tek membranlı organellerdir. Peroksizom, çok uzun zincirli yağ asitlerinin β-oksidasyonu, yağ asitlerinin α-oksidasyonu ve eter-lipitlerin sentezi dahil olmak üzere çeşitli metabolik işlemlerde yer alan 50’den fazla enzimi içeren özel bir enzim “fabrikası” dır. Doğru peroksizom biyogenezinde yer alan ve gerekli olan proteinlere peroksin denir(PEX). İnsanlarda en az 15 PEX geni tanımlanmıştır. PBD-ZSS, peroksin kodlayan 13 PEX geninden birindeki mutasyonlardan kaynaklanır. Bu genlerdeki mutasyonlar, anormal peroksizom biyogenezisine yol açar.
NALD’ın klinik seyri oldukça hızlı
olabilir. Doğumdan sonra psikomotor gelişimin olmaması ve ardından ölüm ile
ilişkilidir. Bazı hastalarda ise, daha az ciddi bir kalıtım gösterir ve
hastalar gençliğinin ortalarına kadar hayatta kalırlar. Bununla birlikte bu
hasalarda sensörinöral sağırlık ve retinopatiye bağlı körlük görülür. Hayatta
kalan hastaların zihinsel gerilik görülür. Gelişimsel gerileme ise,
lökodistrofinin başlaması nedeniyle 3
ila 5 yaşlarında ortaya çıkar.
Belirti ve Semptomlar
NALD doğumla ya da erken
bebeklik dönemlerinde başlar ancak bazen semptomlar geç bebeklik ya da erken
çovukluk dönemlerie kadar teşhis edilemeyecek kadar hafiftir. Hipotoni( gevşek
bebek sendromu, kasın harekete karşı gösterdiği
direnç oldukça düşüktür.), nöbetler, yaygın beyin hasarları, sensörinöral
işitme kaybı, periferik nöropati(el ve ayaklarda zayıflık uyuşukluk), karaciğer
anormalliği, hafif yüz dismorfizmi, gelişememe ve ciddi ölçüde gecikmiş psikomotor
gelişim ile karakterizedir.
Genetik Görülme Sıklığı
PBD-ZSS
(Peroksizom biyogenez bozuklukları) için tahmini doğum yaygınlığı Kuzey
Amerika’da 1 / 50.000 ve Japonya’da 1 / 500.000’dir. PBD-ZSS’li hastaların
yarısından fazlasında NALD-IRD formları vardır.
Kalıtım Paterni/Deseni
Yenidoğan Adrenolökodistrofi, otozomal resesif bir bozukluktur. Otozomal resesif bozuklukların kalıtım paterni figürde gösterilmiştir.
NALD’nin fizik muayenede olduğundan
şüphelenilir ve biyokimyasal değerlendirme ile doğrulanır. Plazma çok uzun
zincirli yağ asidi (VLCFA) konsantrasyonunun analiziyle tespit edilir.
Amniyon sıvıdan alınan örnek ile
doğum öncesi tanı mükündür.
NALD için bir tedavi yoktur ve
tedavi semptomatiktir. Katarakt erken bebeklik döneminde çıkarılmalı ve vizyonu
iyileştirmek için gözlük kullanılmalıdır.İşitme bozukluğu için işitme cihazı
kullanılması gerekebilir. Karaciğer anormalliği için Kvitamini takviyesi
yapılabilir ve Primer safra asidi tedavisi ile karaciğer fonksiyonu
düzelebilir. Yeterli kalori alımını sağlamak için bir gastronomi tüpü
gerekebilir. Nöbetler için standart epileptik ilaçlar kullanılır. İşitme, görme
ve karaciğer fonksiyonlarındaki değişiklikleri izlemek için yaşam boyu takip
gereklidir.
Hastalıkla İlişkili Genler
NALD, peroksizom biyogenezinde yer
alan çeşitli genlerdeki mutasyonlardan kaynaklanabilir (peroksin adı verilen
proteinleri kodlayan PEX genleri). Bu genler :
Akrokeratoelastoidoz, (AKE),
otozomal dominant kalıtılan nadir bir hastalıktır ve ilk kez 1952 yılında
Brezilyalı dermatolog Oswaldo Costa tarafından tanımlanmıştır. 1973 yılında
Jung, ciddi derecede etkilenmiş bir aile ile önemli çalışmalar yapmıştır.
1977’de Matthews ve Harman, anneleri de etkilenmiş iki erkek kardeş üzerinde
çalışmalar yapmışlardır. Hastalık, ellerin ve ayakların iç ve dış kenarlarında
meydana gelen ten rengi lezyonlar, papüller ile karakterizedir. Esas olarak
palmoplantar bölgelerinin yan kısmını etkileyen bir tür marjinal
keratodermadır. Otozomal dominant kalıtımının yanı sıra sporadik vakalar da
bildirilmiştir. Hastalık tipik olarak çocukluk çağında başlar ancak başlangıcı
ergenliğe kadar da gecikebilir, bazı erişkin vakalar da bildirilmiştir. Yaşam
süresi üzerine herhangi bir olumsuz etkisi olmadığından hastalık, estetik
kaygının dışında iyi bir prognoza sahiptir denilebilir.
[1, 2, 3, 6]
Belirti ve Semptomlar
Ellerin ve
ayakların kenarlarında meydana gelen ten rengi küçük 2-4 mm’lik lezyonlar ile
karakterize olmasının yanı sıra hastalıkta eklem üzerleri, tırnak kenarları,
diz, bacağın ön yüzü, ayak ve el bileği ve üst kısımları da etkilenebilir
(Şekil 1,2). 1998’de Fiallo ve
arkadaşları elastoreksinin lezyonların bir özelliği olduğunu bulmuşlardır. Bu
küçük papüller gün geçtikçe bir araya gelerek plaklar oluşturabilir. Lezyonlar, çoğu zaman asemptomatiktir.
Sistematik bir göstergesi belirtilmemiştir. Hiperkeratoz, akantoz, hiyalinize
ve homojen kollajen yapısı, elastik liflerde parçalanma ve azalma hastalığın histolojik
özellikleridir. Etkilenen bölgede hafif kaşıntı, ağrı ve yanma hissi olabilir.
Güneşe maruz kalma veya travma hikayesi lezyonların ortaya çıkışını
teşvik edebildiği biliniyor, özellikle sporadik vakalarda bu maruz kalmalar
veya travmalar dikkate alınmaktadır. El yıkama alışkanlıkları, kıyafetler,
ayakkabı sebepli tahriş gibi durumların AKE’ye sebebiyet verebileceği
bilindiğinden hastalara mutlaka bunlar sorulmaktadır. Zamanla sayıda artış meydana
gelebildiği belirtilmiştir. Hastalığın patogenezi hala tam olarak
bilinmemektedir. Hastalık, ırk ve cinsiyet ayrımı göstermez. Hamilelik
sırasında lezyonların hızlı ilerlediğine dair raporlar olsa da genellikle ilk
birkaç hafta ila ay içerisinde durum stabil hale gelmektedir.
Hastalık, bilateral oluşumlu (yani iki elde de iki
ayakta da gibi) olsa da unilateral, tek taraftaki organda meydana gelmiş
vakalar da bildirilmiştir. Bu durumun, mosaisizm kaynaklı olabileceği
öngörülmüştür.
[1, 2, 3, 4, 5, 6, 7]
(Şekil 1) [2] (Şekil 2) [3]
Hastalığın Genetiği
Sporadik vakaların olmasına rağmen
Akrokeratoelastoidoz’un, birincil olarak otozomal dominant geçiş gösteren
genodermatoz olduğu kabul edilmektedir (Bazı vakaların otozomal resesif geçişli
olduğu da görülmüştür). Stevens ve arkadaşları 1996’da hastalığın otozomal dominant
kalıtıldığını belirtmişlerdir. Palmoplantar keratodermalar (PPK) sınıflandırılırken
akrokeratoelastoidoz (AKE), kalıtsal punktat palmoplanter keratoderma tip III (PPKP3) grubuna dahil edilir. AKE,
PPKP’nin benign yani iyi huylu formudur.
Jung (1973) ve Greiner (1983) önderliğinde yapılan genetik haritalandırma çalışmalarında, AKE’nin ACP1 (Asit fosfataz 1), JK (Kiddi kan grubu) ve IGKC (İmmunoglobulin kappa hafif zincir sabir bölgesi) genleri ile ilişkili olduğu bulunmuştur. Bu üç belirteç gen 2. kromozomun p kolunda (2p) yer alır. Bu sebeple hastalığın lokusunun 2p olduğu düşünülmektedir. Kromozom 2’yi içeren bu genetik anormallik ile genden eksprese olan protein kompleksinde meydana gelen değişikliğin hastalığın sebebi olabileceği ileri sürülmüştür. Yine de bunun hakkında fazla çalışma bulunmamaktadır.
(Şekil
3) [11]
Otozomal dominant kalıtımda (Şekil
3) ebeveynlerden bir tanesi bile hasta ise çocuklarında %50 veya %100 oranında
etkilenme olasılığı vardır (Hasta ebeveynin heterozigot veya homozigot oluşuna
bağlı olarak). Resesifteki gibi taşıyıcı olma durumu yoktur. Sporadik vakalarda
ise ailede, ebeveynlerde hastalık olmasa bile kişide görülebilir.
[1,
6, 11]
Tanı
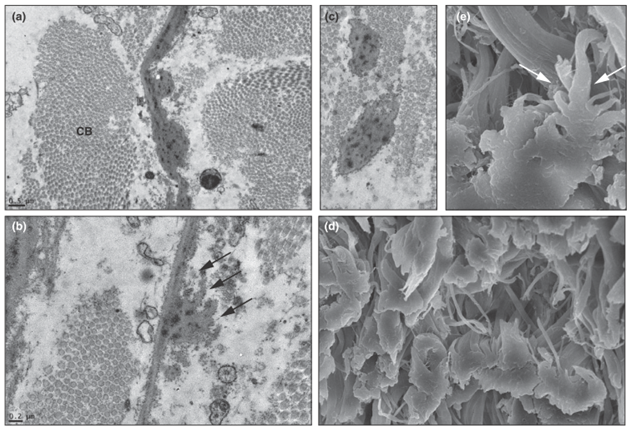
Sporadik veya kalıtsal AKE formları arasında mikroskobik ve klinik farklar yoktur. Nadir bir hastalık olduğundan mevcut elektron mikroskop görüntüleri sınırlıdır. Elektron mikroskopisi, elastik liflerin mikrofibriler fragmantasyon ile ayrışmasını gösterir.
(Şekil 4) [5]
Elektron
mikroskopisi inceliği azalan elastik fiberleri kolaylıkla gösterebilir.
Şekil
4’te bir hastanın transmisyon elektron mikroskopi (TEM) ve taramalı elektron
mikroskopi (SEM) görüntüleri mevcuttur. Buna göre a, b ve c TEM; d ve e SEM
görüntüleridir.
Normal
kollajen demeti olan ince elastik fiber (CB: collagen bundle) (x 20.000)
Yüzey
girintileri olan ince elastik fiber, oklarla gösterilmiş (x 30.000)
Horoz
tepesine benzeyen girintili elastik fiberler, oklarla gösterilmiş (x 3.700)
Hastalığın
klinik olarak teşhisinde daha önce belirtildiği gibi el ve ayakların iç ve dış
kenarlarında mevcut ten rengi veya sarı renkli lezyonlar baz alınır. Hiperkeratoz,
akantoz, hiyalinize ve homojen kollajen yapısı, elastik liflerde parçalanma ve
azalma hastalığın histolojik özellikleridir. Etkilenen bölgede hafif kaşıntı,
ağrı ve yanma hissi olabilir.
AKE ,
diğer papüller akrokeratoderma, ksantomlar, düz siğiller, piezojenik pedal
papülleri vb gibi olgulara da benzediğinden tanısının konulması çok kolay
olmayabilir. [5, 7, 10]
Tedavi
Lezyonlar
genellikle asemptomatik olduğundan tedavi ertelenebilir. Ancak hastalarda
özellikle estetik açıdan kaygılar mevcuttur. Hasta, durumun doğası ve sınırlı
tedavi yöntemleri hakkında bilgilendirilmelidir.
Salisilik
asit, üre, sülfür, katran ve tretinoin gibi topikal keratolitikler yani deri
üzerinden uygulanan tedaviler önerilebilir. Prednizolon, antibiyotikler,
izotretinoin, dapson, metotreksat ve asitretin gibi oral yolla alınan immünsupresifler
ile sistemik tedavi de hastalıkta uygulanabilir. Bu tedavi yaklaşımları başarı
sağlamaktadır.
Kriyocerrahi,
hastalıkta denenmiştir ama yıkıcı etkilerinden dolayı tavsiye edilen bir yöntem
olmamıştır.
Nelson-Adesokan
ve arkadaşları, 50 mg/gün oral etretinat ile geçici bir klinik yanıt elde
ederek lezyonların giderilmesini sağlamışlar ancak ilaç kullanımı kesildikten
sonra lezyonların geri geldiği görülmüştür.
Erbium:
YAG lazer operasyonunun tedavide kullanımı denenmiştir [10]. Bölgesel anestezi
ile uygulanan bir yöntemdir. Lazer sonrası oluşan yaralar ağrısız ve herhangi
bir komplikasyon olmadan iyileşmektedir. Lezyonların hafif şekilde giderildiği
ve düzleştiği görülmüştür. Altı ay boyunca yapılan takipte nüks etmediği
görülmüştür. Hastalığın kısmi şekilde giderilmesi ile önerilen bir tedavi
olmaktadır ama tamamen bir iyileşme beklenmez.
AKE’nin
tedavisi hala tartışma konusudur.
[1,
8, 9]
Hastalığın Diğer İsimleri
Acrokeratoelastoidosis (AKE) Of Oswaldo
Costa,
Inverse Papular
Acrokeratosis,
Collagenous Plaques Of Hand And Feet,
Palmoplantar
Keratoderma, Punktat Tip III; PPKP3
Lichenoid Akrokeratoelastoidosis
Kaynaklar
SONTHALIA, S.,
ABOOBACKER, S., 2019, Acrokeratoelastoidosis, Stat Pearls.
ERBİL, A.H., SEZER, E., KOÇ, E., TUNCA, M.,
TAŞTAN, H.B., DEMİRİZ, M., 2007, Acrokeratoelastoidosis treated with the
erbium: YAG laser, Clinical and Experimental Dermatology, 33(1), 30-31.