Belirti
ve Semptomlar
WWS’nin ana semptomları kas distrofisi , beyin ve göz
anormallikleridir. WWS semptomları konjenitaldir (doğumda bulunur) ve bazı
beyin anormallikleri gebeliğin sonraki aşamalarında prenatal ultrason ve / veya
fetal MRI ile tespit edilebilir.
WWS’li bireylerde konjenital kas distrofisi veya doğumda kas
zayıflaması ve kaybı vardır. Kas distrofisi, etkilenen bebeklerin “disket
bebek” sendromu olarak belirtilebilecek ciddi hipotoni (düşük kas tonusu)
olmasına neden olur. Kas zayıflığı ve atrofi (israf) tipik olarak zamanla
kötüleşir. Etkilenen bazı kişiler kas lifleri gibi doku kalınlaşması ve
kısalması sırasında oluşan anormal olarak sabit eklemler (kontraktürler)
geliştirir, etkilenen bir alanın hareketini deforme eder ve kısıtlar.
Etkilenen bebekler genellikle lissensefali (düz beyin),
hidrosefali (genişlemiş ventriküller) ve beynin arkasındaki malformasyonlar
dahil olmak üzere çeşitli ciddi beyin bulgularına sahiptir. WWS’ye dahil olan
lisensefali tipi, tip 2 veya parke taşı lisensefali olarak tarif edilir. Bunun
nedeni, beynin yüzeyinin pürüzsüz olması ve yüzeydeki beyin hücrelerinin
(nöronların) kümelerinin toplanması nedeniyle bir parke taşı görünümüne sahip
olmasıdır. Beynin ventriküllerinde genişlemeye neden olan çok fazla beyin omurilik
sıvısı olması ile karakterize edilen hidrosefali, oldukça şiddetli olabilir ve
anormal derecede büyük bir kafaya yol açabilir. Beynin arka bölümlerinin
malformasyonları, beyincik ve beyin sapının az gelişmişliğini (hipoplazi)
içerebilir. Beyincik, istemli kas hareketlerini koordine etmeye yardımcı olur;
beyin sapı ise solunum, tükürük salgılama ve kalp atış hızı gibi temel
fonksiyonları kontrol etmeye yardımcı olur. Bu posterior malformasyonlar, bazen
Dandy-Walker malformasyonu olarak da adlandırılan beynin arkasında anormal
olarak genişlemiş bir alanı içerebilir. WWS’li bazı bireylerde, beynin bir
kısmının kafatası kemiğinden (ensefalosel) ve / veya normal olarak iki beyin
yarıküresini birbirine bağlayan beyaz madde bandı olan corpus callosum yoktur.
WWS ile ilişkili göz (oküler) anormallikleri kişiden kişiye
değişir ve aşağıdakilerden herhangi birini içerebilir: anormal derecede küçük
gözler (mikroftalmi), eksik veya az gelişmiş optik sinirler (optik sinir
hipoplazisi), sıvı içindeki dolu alanların malformasyonları gözler korneanın
arkasında ve irisin önünde ve retinanın ayrılmasına neden olabilecek retinanın
malformasyonu (retinal displazi). Ek göz semptomları arasında katarakt, yarık retina
veya irisin doku kaybı (kolobomlar), anormal derecede büyük ve çıkıntılı gözler
(buptalmi) veya gözlerde artan basınç (glokom) sayılabilir. Bu anormalliklerin
çoğu kısmi veya tam körlüğe neden olur.
WWS ile ilişkili beyin malformasyonları bebeklik döneminde
ciddi, hayatı tehdit eden komplikasyonlara neden olur. WWS ile doğan çocuklar
çeşitli derecelerde zihinsel engellilik gösterir ve sıklıkla nöbet geçirir.
Birleştirilmiş beyin ve kas anormallikleri, gelişimsel kilometre taşlarına
ulaşmada önemli gecikmelere yol açar (örn. Oturma, nesneleri yakalama, tarama,
konuşma) ve nefes alma, yutmada zorluklara neden olacak kadar şiddetli
olabilir. Bazen, farklı vücut sistemlerinde ek semptomlar da mevcut olabilir.
Etkilenen bazı çocuklarda, üriner sistem blokajı ve böbrek pelvik dilatasyonuna
(hidronefroz) veya testislerin erkeklerde skrotuma inmemesine (kriptorşidizm)
neden olan genitoüriner anormallikler oluşabilir. Etkilenen bazı çocuklar,
düşük ayarlı veya belirgin kulaklar, yarık damak veya yarık dudak gibi başka
özelliklere sahip olabilir.
Nedenler
WWS, beyin, göz ve kas gelişiminde önemli olan anormal
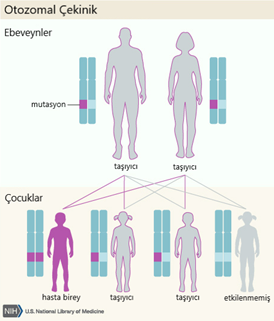
şekilde çalışan veya çalışmayan genlerden kaynaklanır. Otozomal resesif bir
şekilde kalıtsaldır ve bu nedenle her ebeveynden bir gen olmak üzere bir genin
iki anormal kopyasını miras alan bir bireyde ortaya çıkar. Genin normal olarak
çalışan bir kopyasına ve genin çalışmayan bir kopyasına sahip olan bir birey
WWS için bir taşıyıcıdır, ancak genellikle herhangi bir semptomu yoktur.
Birlikte çocuğu olan iki taşıyıcı anne-babanın anormal veya çalışmayan geni
geçmesi ve bu nedenle etkilenen bir çocuğu olması riski, her hamilelikte% 25
veya 4’te 1’dir. Bu ebeveynlerin her hamilelikte sadece taşıyıcı (etkilenmemiş)
bir çocuk sahibi olma riski% 50 veya 2’de 1’dir. Genlerin normal olarak işleyen
iki kopyası olan (etkilenmeyen ve taşıyıcı olmayan) bir çocuğa sahip olma
şansları, her hamilelikte% 25, 4’te 1’dir. Bu riskler, erkek ve dişi yavrular
için aynıdır.
WWS, beyin, göz ve kasın düzgün gelişimi ve işlevinde önemli
bir rol oynayan spesifik proteinlerin eksikliklerine veya tam eksikliğine neden
olur. Bu proteinler, kas hücrelerinin ve sinir hücrelerinin zarlarında bulunan
α-dystroglycan adı verilen başka bir protein ile birlikte işlev görür. α -dystroglycan
normalde kas hücrelerini stabilize etmek ve gelişim sırasında beyindeki sinir
hücrelerinin göçüne yardımcı olmak için çalışır. WWS ile ilgili proteinler,
şeker moleküllerinin α-dystroglycan’a bağlanması için, α-dystroglycan’ın düzgün
çalışması için gerekli olan glikosilasyon adı verilen bir işlemde gereklidir.
WWS’li bireylerde, glikosilasyona dahil olan proteinlerden birini kodlayan
genler mutasyona uğrar ve bu nedenle α -distroglikan düzgün glikosile edilmez
ve normal işlevlerini yerine getiremez. α -distroglikan glikosilasyondaki
bozukluklar WWS’nin ve ilgili distroglikanopatilerin beyin, göz ve kas
özelliklerine yol açar.
WWS, proteinleri normalde yukarıda tarif edilen
glikosilasyon işlemine dahil etmekten sorumlu olan en az 14 farklı gene
bağlanmıştır. Alfabetik olarak listelenmiş, şimdiye kadar tanımlanan genler
şunlardır:
• B3GALNT2: Beta-1,3-N-asetilgalaktozaminiltransferaz 2
proteini
• B4GAT1: Beta-1,4-glukuroniltransferaz 1 proteini
• DAG1: Distrofin ilişkili glikoprotein 1
• FKRP: Fukutinle ilişkili protein
• FKTN: Fukutin proteini
• GMPPB: GDP-mannoz pirofosforilaz B proteini
• ISPD: İzoprenoid sentaz domenini içeren protein
• BÜYÜK: Asetilglukosaminiltransferaz benzeri protein
• POMT1: O-mannosiltransferaz 1 proteini
• POMT2: O-mannosiltransferaz 2 proteini
• POMGNT1: O-mannoz beta-1,2-N-asetilglukosaminiltransferaz
proteini
• POMGNT2: O-mannoz beta-1,4-N-asetilglukosaminiltransferaz
2 proteini
• POMK: Protein-O-mannoz kinaz
• TMEM5: Transmembran proteini 5
Yukarıda belirtilen 14 gen WWS’nin nedenleri olarak
tanımlanmış olsa da, bilinen WWS vakalarının sadece yarısını açıklarlar ve tüm
bu genlerdeki mutasyonlar da daha az şiddetli distroglikanopat formlarına neden
olabilir. WWS’nin bilinen sayısız genetik nedenine rağmen, genetik test her
ailede nedensel geni ortaya çıkarmayabilir. Bu nedenle, gelecekte WWS ve ilgili
koşullarla ilişkili daha fazla gen keşfedilecek ve bu da WWS spektrumuna ek
değişkenlik getirebilir.
Etkilenen
Popülasyonlar
WWS dünya çapında rapor edilmiştir. Erkekleri ve kadınları
eşit sayıda etkilemektedir. İnsidansı bilinmemektedir, ancak 200.000’de 1’den
az olduğu tahmin edilmektedir.
İlgili
Bozukluklar
Birkaç bozukluğun belirtileri WWS ile örtüşebilir. CMD
birkaç farklı durum içeren bir grup kas hastalığı için genel bir terimdir. WWS
gibi bazı CMD formları yapısal beyin malformasyonları ve zihinsel engellilik
ile ilişkili olabilirken, diğerleri genellikle merkezi sinir sistemini içermez.
Bu bozuklukların şiddeti, spesifik semptomları ve ilerlemesi büyük ölçüde
değişir. CMD’nin hemen hemen tüm bilinen formları otozomal resesif koşullar
olarak kalıtsaldır. CMD’nin iki spesifik formu, Fukuyama CMD ve kas-göz-beyin
hastalığı (MEB) WWS’ye benzer semptomlara sahiptir, ancak bunlar genellikle
daha az şiddetlidir. WWS ile ilgili CMD’ler, beyin kistleri veya beyin
malformasyonları olmadan zihinsel sakatlık gibi daha hafif beyin fenotipleri
ile de ortaya çıkabilir.
Tip II lisensefali de zaman zaman kas distrofisi olmadan
izolasyonda gözlenmiştir, ancak bu vakalarda genetik nedenleri bilinmemektedir.
İzole tip II lisensefali’nin WWS ile ilişkili olup olmadığı belirsizdir. Tip I
lissencephaly farklı bir durum gibi görünmektedir.
Teşhis
WWS tanısı, gebeliğin geç evrelerinde rutin ultrason ve /
veya fetal MRI ile şüphelenilebilir ve çeşitli klinik testlerin yapılmasını ve
çeşitli testler gerektirebilecek karakteristik bulguların tanımlanmasını
takiben doğumda veya doğumdan kısa bir süre sonra doğrulanabilir.
Beyin bulgularının tanımlanması en iyi, beynin ayrıntılı
resimlerini sağlayan manyetik rezonans görüntüleme (MRI) kullanılarak yapılır.
Bununla birlikte, sıklıkla WWS tanısına eşlik eden genişlemiş ventriküller,
başın ultrason ve bilgisayarlı tomografisi (BT) ile de tespit edilebilir. Kas
liflerindeki karakteristik değişiklikleri ortaya çıkarmak için kas dokusunun
biyopsi ve mikroskopik olarak değerlendirilmesi gerekebilir. CK, kas dokusunun
parçalanmasının bir ölçüsü olduğundan, kreatin kinaz (CK) seviyeleri için bir
kan testi de yaygın olarak yapılır. Yüksek CK seviyelerinin tespiti, kasın
hasar gördüğünü veya iltihaplandığını doğrulayabilir, ancak özellikle WWS
tanısını doğrulayamaz. Dikkatli bir göz muayenesi, WWS’nin karakteristik göz
bulgularını da belirleyebilir.
Genetik test, WWS’nin klinik teşhisinin moleküler
doğrulamasını sağlamak için yapılabilir. Genetik doğrulama, bilinen bir
nedensel gende iki mutasyon tanımlandığında meydana gelir. Genetik test çeşitli
yollarla yapılabilir. Genetik bir test yöntemi, bilinen tek bir WWS geninin
dizilimidir ve belirli bir genden şüpheleniliyorsa yapılabilir. Bilinen çok
sayıda WWS veya CMD geninin bir kerede çok genli bir panel testi ile test
edilmesi de mümkündür. Başka bir genetik test yöntemine, bilinen her genin bir
defada mutasyonlar için analiz edilebildiği tüm ekzom dizilemesi denir. Bu
yöntemlerin her birinin faydaları ve sınırlamaları vardır ve bireysel koşullar
bir yöntemi diğerine göre önerebilir. Etkilenen kişi Ashkenazi Yahudi kökenli
ise, önce FKTN geni test edilmelidir, çünkü bu gende spesifik bir mutasyon bu
popülasyonda yaygındır. Aksi takdirde, bilinen tüm WWS genlerinde mutasyonların
neden olduğu semptomların önemli ölçüde çakışması vardır, bu nedenle genetik
test için belirli bir düzen belirlemek zordur.
WWS için tüm genlerin henüz tanımlanmamış olması muhtemel
olduğundan, genetik test negatif olabilir. Bu nedenle, negatif bir sonuç,
öncelikle klinik semptomlara dayanarak yapılan WWS tanısını dışlamaz ve bu
durumlarda, genetik test fırsatlarının (yeni tanımlanan WWS genlerini içerecek
şekilde) yeniden değerlendirilmesi gelecekte periyodik olarak düşünülebilir.
Aileler için genetik danışmanlık, otozomal resesif kalıtım,
WWS için mevcut ve sürekli değişen genetik testin durumu, bir aile için genetik
testin sonuçlarının ne anlama geldiği ve bu bilgilerin ailedeki bireyler
üzerindeki etkileri konusunda anlayışlarına yardımcı olabilir.
Tedavi
Şu anda WWS için bir tedavi yoktur ve tedavi spesifik
semptomlara göre bireyselleştirilir. Tıbbi yönetim, çocuk doktorları,
genetikçiler, ortopedi cerrahları, nörologlar, göz uzmanları ve diğer sağlık
uzmanları da dahil olmak üzere bir uzman ekibinin etkilenen bir çocuğun
tedavisini sistematik ve kapsamlı bir şekilde planlaması için koordineli
çabalarını gerektirebilir.
Tedaviler, anti-nöbet ilacı, aşırı beyin omurilik sıvısını
boşaltmak ve basıncı azaltmak için şantların implantasyonu gibi hidrosefali
ameliyatı ve kas gücünü iyileştirmek ve kontraktürleri önlemek için fizik
tedaviyi içerebilir. Etkilenen bazı bebekler, beslenme güçlüklerine yardımcı
olmak için bir mide tüpüne ihtiyaç duyabilir. Diğer semptomatik ve destekleyici
tedaviler de gerekli olabilir. Şiddetli beyin ve kas anormallikleri nedeniyle,
klasik WWS’li çocukların yaşam beklentileri her zaman azalır.
Diğer
İsimler:
- Hidrosefali
- Agriya ve retina displazisi
- Sert sendrom
- Sert +/- E sendromu
- Warburg sendromu
- Chemke sendromu
- Pagon sendromu
- Serebrooküler disgenez
- Serebrooküler displazi kas distrofisi sendromu
- COD-MD sendromu
Kategoriler:
- Konjenital ve Genetik Hastalıklar
- Göz hastalıkları
- Metabolik bozukluklar
- Sinir Sistemi Hastalıkları
Bu
hastalık aşağıdaki gruplara ayrılır: Gelişimsel anomali ile
konjenital glikosilasyon bozukluğu; Beyin ve göz anomalileri ile konjenital kas
alfa-distrokancanopati; Konjenital müsküler distrofi; Distroglikanopatiye bağlı
konjenital müsküler distrofi
Özet
Walker-Warburg sendromu (WWS), beyin ve göz anormallikleri
ile ilişkili ciddi bir konjenital kas distrofisidir. Belirti ve semptomlar
tipik olarak doğumda bulunur ve hipotoni, kas güçsüzlüğü, gelişimsel gecikme,
zihinsel sakatlık ve nadiren nöbetleri içerir. Ayrıca lisensefali, hidrosefali,
serebellar malformasyonlar, göz anormallikleri ve diğer anormallikler ile
ilişkilidir. Çoğu çocuk üç yaşından sonra hayatta kalmaz. Birçok kişide genetik
neden bilinmemekle birlikte, POMT1, POMT2 ve FKRP genleri dahil olmak üzere
birçok genin mutasyonlarından kaynaklanabilir. WWS otozomal resesif bir şekilde
kalıtsaldır. Özel bir tedavi mevcut değildir; genellikle destekleyici ve
önleyicidir.
Referanslar
{kind=link}