Fumarik asitüri,beyni ve sinir sisteminin diğer kısımlarını etkileyen kalıtımsal bir hastalıktır.Belirti ve semptomlar küçük kafa(mikrosefali),şiddetli gelişimsel gecikme,zayıf beslenme,zayıf kas tonusu(hipotoni),gelişememe,nöbetler ve ayırt edici yüz özellikleri olabilir.Maalesef,bu eksikliğe sahip kişilerin çoğunun konuşma ve yürüme yeteneği yoktur.Çeşitli beyin anomalilerinin tespiti MRI’da tespit edilebilir.Fumaraz eksikliği FH genindeki mutasyondan kaynaklanır ve otozomal resesif olarak kalıtılır.FH geninin ürünü olan fumarat hidrataz Krebs Döngüsü’nde fumaratın malata dönüşümünü katalizleyen enzimdir.Maalesef şu anda etkili bir tedavi yoktur.
Genetik Değişiklikler
Fumaraz eksikliği FH genindeki mutasyonan kaynaklanır ve mutasyonun şiddetine göre risk değişmektedir.Bu gen vücuda fumarat hidrataz üretimini sağlar.Bu enzim ise hücrelere oksijeni kullanarak enerji üretmede yardımcı olur.Gendeki mutasyon enzimin çalışmasını engeller.Enerji üretimi özellikle beyin gelişimi boyunca oldukça önemlidir.
Belirti ve Semptomlar
Fumaraz
eksikliği olan çoğu yenidoğan yetersiz beslenme.gelişememe ve zayıf kas
tonusu(hipotani) gibi bazı nörolojik anomalilere sahiptir.Erken başlangıç
infantil ensefalopatisi(beyin yapısı veya fonksiyonunda
anormallikler),nöbetler,mikrosefali ve geç gelişim de görülmektedir.Fumaraz
eksikliği olan çoğu çocuk konuşmayı ve yürümeyi hiç öğrenemez.Bazılarında
ayrıca polisitemi(kırmızı kan hücresi fazlalığı),tekrarlayan
kusmalar,pankreatit görülmüştür.Ayırt edici yüz özellikleri arasında öne çıkmış
alın,baskılanmış burun köprüsü ve ayrık gözler bulunabilir.
MRI ile çeşitli beyin anomalileri tespit edilebilir.Bunlar arasında serebral atrofi(nöronların ve birbirleriyle bağlantılarının kaybı),genişlemiş ventriküller(beyin omurilik sıvısının üretildiği boşluklar),korpus kallozomun incelmesi veya yokluğu(agenez),anormal derecede küçük beyin sapı vardır.İdrarda yüksek aminoasit seviyesi(aminoaidüri) de önemli semptomlardandır.
Genetik Görülme Sıklığı
Fumaraz eksikliği 2017 yılı itibariyle yaklaşık 100 raporlu vaka ile nadir bir hastalıktır.Hastalık farklı etnik kökene sahip insanlarda oluşur ama bu hastalığa sahi birçok kişi güneybatı Amerika’nın(Kuzey Arizona ve Güney Utah) izole edilmiş bölgelerindeki topluluklardadır. Buradaki artan sıklığın akraba evliliklerinden ve çok eşi topluluklardan kaynaklandığı düşünülüyor.Genetik görülme sıklığı ise <1/1000000’dur.
Kalıtım Paterni
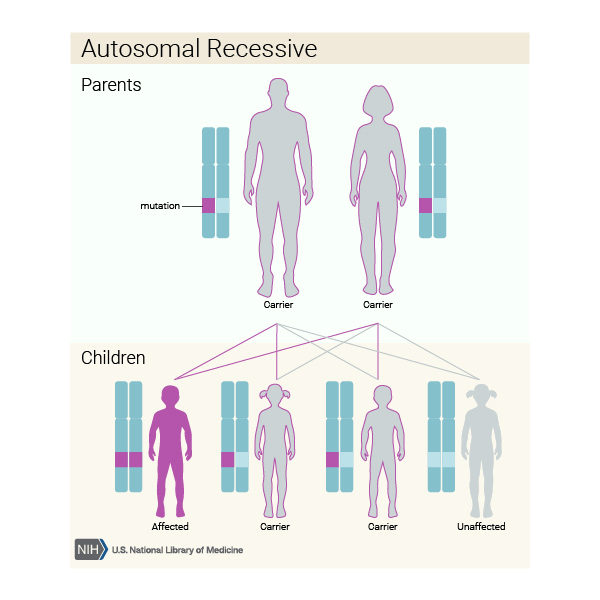
Otozomal resesif kalıtılır.Bunun anlamı mutasyona uğramış genin eşey hücrelerinde değil vücut hücrelerinde kalıtıldığı yani cinsiyetten bağımsız olduğudur.
Teşhis Yöntemleri
Organik
asitlerin kromatografisi, sıklıkla süksinik asit ve alfatotoglutarik asit ile
ilişkili, atılan fumarik asit kanıtı sağlar. Hiperlaktakidemi ve orta derecede
hiperammonemi diğer yaygın bulgulardır. Tanı, lökositlerde veya kültürlenmiş
fibroblastlarda fumarat hidrataz aktivitesini ölçerek doğrulanabilir. Beyin
MRI’sı serebral atrofi, genişlemiş ventriküller ve genişlemiş ekstra eksenel
serebral omurilik sıvısı (BOS) boşlukları, yaş için miyelinasyonun gecikmesi,
korpus kallozumun incelmesi ve anormal derecede küçük bir beyin sapı gibi
çeşitli anomalileri ortaya çıkarır. Bilateral polimikrojiyi ve korpus
kallozumun yokluğunu içeren gelişimsel malformasyonlar da görülebilir.
Ayırıcı Tanı
İdrardaki yüksek
fumarik asit seviyesi metabolik stresden kaynaklanabilir; bu nedenle fumarik
asitüri testi hasta stabilize edildikten sonra tekrarlanmalıdır. Ayırıcı
tanılara polimikroji ve Leigh sendromu dahildir.
Doğum Öncesi Tanı
Polihidramnios,
intrauterin büyüme gecikmesi ve erken doğum vakaların üçte birinden fazlasında
görülür. Fetal ultrason genişlemiş serebral ventrikülleri ve diğer beyin
anormalliklerini ortaya çıkarır.
Tedavi
FA hastalarına
sadece semptomatik tedavi uygulanır. Yenidoğanlarda beslenmeyi kolaylaştırmak
için gastrostomi gerekebilir. Nöbetleri kontrol etmek için kullanılan
terapiler, bu enzimatik kusur ailesi için kontrendike olan ketojenik bir diyet
içermemelidir. Skolyozu azaltmak ve hareketliliği artırmak için fizik tedavi
bazı durumlarda yardımcı olabilir. Daha az etkilenen vakalarda motor becerileri
ve dil gelişimini geliştirmek için özel eğitim ve mesleki terapi gereklidir.
Uzun süreli hayatta kalan hastalar için düzenli tümör testi gereklidir.
B12 Vitamini
duyarlı metimalonik Asidemi, vücudun belirli proteinleri ve yağları
parçalayamadığı kalıtsal bir durumu ifade eder. Bu durumun sonucu olarak
vücutta toksik maddelerin birikmesi, dekompasyon (tıpta bir organın vücut
ihtiyaçlarını karşılayamaması anlamına gelir) oluşmasına veya ciddi hastalık
nöbetlerine yol açar. Genellikle bebeklik döneminde ortaya çıkar ve etkileri
çok hafiften hayati dereceye kadar uzanabilir. Uzun süreli komplikasyonlar
arasında büyüme gecikmesi, zihinsel yetersizlik, böbrek hastalığı ve pankreatit
sayılabilir. Metimalonik asidemi, birkaç farklı gendeki değişikliklerden
kaynaklanır ve otozomal resesif olarak kalıtsaldır. Tedavisi için düşük
proteinli bir diyet, ilaçlar, antibiyotikler ve bazı durumlarda karaciğer ve
böbrek nakli içerir. Tedavisi olmazsa, bazı durumlarda komaya ve ölüme yol
açabilir.
Klinik Tanım
Metilmalonik
asidemiler, dört amino asidin (metiyonin, treonin, izolösin ve valin)
metabolizmasında enzimatik bir kusurun neden olduğu organik asidemilerdir. Hastalarda
genellikle bebeklik veya erken çocukluk döneminde uyuşukluk, gelişim bozukluğu,
tekrarlayan kusmalar, dehidrasyon, solunum sıkıntısı, kas hipotonisi (kasın
harekete karşı gösterdiği direnç), hepatomegali(karaciğer büyümesi) ve koma
gibi özellikler gözlenir. Ayrıca beyin sapını etkileyen metabolik inme ile
birlikte anemi (megaloblastik değil) belirtileri gösterebilir, potansiyel
olarak hayatı tehdit eden ketoasidoz ve / veya hiperammonemi(kanda amonyak
düzeyinin artması) ve gelişimsel gecikme ve entelektüel eksiklik(zeka geriliği)
gösterebilirler. Metilmalonik asidemi sıklıkla ergenlik veya yetişkinlik
nedeniyle son dönem böbrek yetmezliğine yol açar.
Bozukluğa metilmalonil CoA mutaz, metilmalonil rasemaz veya
adenosilkobalamin sentetik enzimlerin bir veya daha fazlasının eksikliği neden
olabilir. İdrarda bir amino asit metabolizması ürünü olan metilmalonatın
atılımı anormal derecede yüksektir ve bu nedenle bozukluğun bir belirtecidir.
Ayrıca B12
vitaminine duyarlı metilmalonik asidemi, adenosilcobalamin (AdoCbl)
sentezindeki kusurlardan kaynaklanır. Üç farklı çeşidi vardır ve bunlar kobalamin
A (cblA), kobalamin B (cblB) ve kobalamin Dv2 (cblDv2)’dir. CblB hastaları
genellikle, cblA hastalarından daha ciddi şekilde etkilenir.
Kobalamin A
MMAA geninde mutasyonu olması sonucu oluşur. Otozomal resesif
olarak kalıtılır. Metilmalonik asidemili hastaların yaklaşık %25’ini bu grup
oluşturur. mut- ‘e benzer klinik görülür ve B12 tedavisine yanıt verir.
Kobalamin B
MMAB geninde
mutasyon olması sonucu oluşur. Otozomal resesif olarak kalıtılır. İzole
metilmalonik asidemili hastaların yaklaşık %12’sini bu grup oluşturur. Klinik
tablo mut0 fenotipine benzer ve B12 tedavisine yanıt verebilir.
Nedenleri – Bu
Hastalık Nasıl Oluşur?- Etoloji
MMUT, MMAA,
MMAB, MMADHC ve MCEE genlerindeki mutasyonlarla metimalonik asidemi ortaya
çıkmaktadır. Bu genlerden hangisinin mutasyona uğradığı ve uğradığı mutasyonun
etkisine göre hastalığın uzun vadeli etkileri değişebilmektedir.
MMUT genindeki mutasyon metimalonik asidemi
hastalığına yol açan mutasyonlar arasında %60 oranda gözükmektedir. Bu genin
görevi CoA(mutaz) adı verilen bir enzimin yapılması için talimat vermektir. Bu
enzim ise bazı protein yapıtaşlarını (amino asitleri), belirli lipitleri ve
kolestrolü parçalamak için B12 vitamini kullanır. MMUT genindeki mutasyonlar,
enzimin yapısını değiştirir veya bu moleküllerin düzgün bir şekilde
parçalanmasını sağlayan enzim miktarını azaltır. Sonuç olarak, metilmalonil CoA
adı verilen bir madde ve diğer potansiyel olarak toksik bileşikler vücudun
organlarında ve dokularında birikerek metilmalonik asideminin belirti ve
semptomlarına neden olabilir.
MMUT genindeki
mutasyonlar herhangi bir enzimin üretimini önlüyorsa bu durum mut0 olarak
adlandırlır ve bu durum metilmalonik asideminin en şiddetli şeklidir ve en kötü
sonuca sahiptir. Diğer bir durum ise Metilmalonil CoA mutaz yapısını değiştiren
ancak aktivitesini ortadan kaldırmayan mutasyonlardır ve mut- olarak belirtilir.
Bu durum tipik olarak mut0 formundan daha değişken ve az şiddetli semptomlara
sahiptir.
Belirtildiği
gibi bazı diğer genler (MMAA, MMAB, ve ya MMADHC) de mutasyona neden olur.
MMAA, MMAB ve MMADHC genlerinden üretilen proteinler, metilmalonil CoA mutazın
düzgün çalışması için gereklidir. Bu üç genden üretilen proteinleri etkileyen
mutasyonlar, metilmalonil CoA mutaz aktivitesini bozarak metilmalonik asidemiye
yol açabilir.
Birkaç başka
metilmalonik asidemi vakasına MCEE genindeki mutasyonlar neden olur. Bu genin
görevi meilmalonil CoA epimeraz adı verilen enzimin üretilmesi için talimat
verilmesidir. Bu enzimde tıpkı Metilmalonil CoA Mutaz gibi bazı amino
asitlerin, lipitlerin ve kolestrolün parçalanmasında rol oynar. Metilmalonil
CoA epimerazının işlevindeki bozulma, hafif bir metilmalonik asidemi formuna yol
açar.
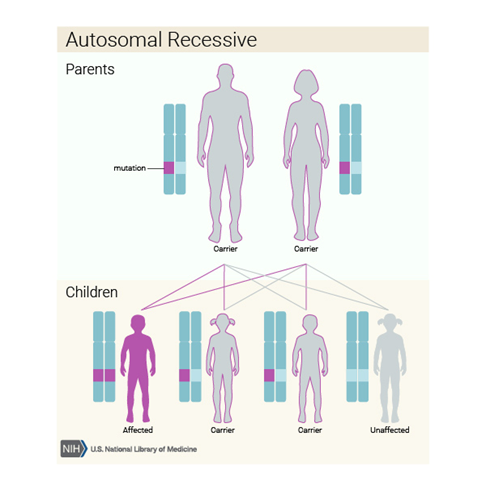
Bu durum otozomal resesif bir paternde kalıtsaldır, yani her
hücredeki MMUT, MMAA, MMAB, MMADHC veya MCEE geninin her iki kopyasında
mutasyonlar vardır. Çoğu zaman, otozomal resesif durumu olan bir bireyin
ebeveynleri mutasyona uğramış genin bir kopyasının taşıyıcılarıdır, ancak
durumun belirtilerini ve semptomlarını göstermezler.
Semptomlar
Metilmalonik
asidemi, kanda toksik maddelerin birikmesinden kaynaklanan dekompasyon olayları
olarak adlandırılan bazı hastalık ataklarına neden olur. Dekompansasyon
olaylarının tedavisinde gecikilirse beyin hasarına yol açabilir. Bir
dekompansasyon olayının belirtileri doğumdan birkaç gün sonra ortaya çıkar ve
şunları içerebilir; yetersiz beslenme ve iştahsızlık, kusma, zayıf kas tonusu
(hipotoni), uyuşukluk (enerji eksikliği), nöbetler, koma ve ciddi diğer
komplikasyonlar genişlemiş karaciğer, entelektüel ve motor sakatlık, kötü
büyüme, böbrek hastalığı ve böbrek yetmezliği ayrıca görüş problemlerini de
içerebilir. Metilmalonik asideminin bazı alt tiplerinde spesifik semptomlar
görülebilir. Bunlara bir örnek olarak homosistinüri ile birlikte metilmalonik
asidemi alışılmadık derecede küçük bir kafa olmasına neden olabilir.
Epidemeyolojisi
Dünyada yaklaşık 1/80,000 – 1/100,000 bebek metilmalonik asidemi ile doğar. Amerika Birleşik Devletleri’nde bir çalışma, yaklaşık 1/90.000 bebeğin metilmalonik asidemi ile doğduğunu tahmin etmiştir.
Resimden de görüldüğü üzere taşıyıcı anne ve babanın her çocuğunda ¼ oranda gözükme olasılığı bulunmaktadır.
Teşhis
Metilmalonik asidemi yeni doğan taraması ile teşhis edilebilir. Ayrıca tanı, kan ve idrardaki artmış metilmalonik aside dayanır. Kurutulmuş kan lekelerinde tandem kütle spektrometrisi (MS / MS) ile propionilkarnitin ve / veya artan propionilkarnitin-asetilkarnitin oranı için yenidoğan taraması yaygın hale gelmiştir, ancak metilmalonik asidin spesifik tanımlanması önemini korumaktadır.
Ayırıcı tanılar arasında, megaloblastik anemi veya homosistinüri olmadan B12-tepkisiz metilmalonik asidemi vitamini varlığı ile ayırt edilebilen, cblC, D ve F’deki kusurların neden olduğu homosistinüri ile metilmalonik asidemi bulunur. Benzer semptomlarla yaşamın erken dönemlerinde (<1 ila 4 hafta) görülür. Tamamlama analizi, etkilenen geni tanımlamak için ilgili grubu veya nedensel genlerin dizilimini tanımlamak için kullanılabilir.
Doğum öncesi tanı, amniyotik sıvıdaki metilmalonat ve trimester ortalarında maternal idrar ölçümü ve kültürlenmiş amniyotik sıvı hücrelerinde fonksiyonel mutaz aktivitesi ve kobalamin metabolizması çalışmaları ile mümkündür. Etkilenen gen ve ailedeki mutasyon (lar) biliniyorsa moleküler tanı mümkündür.
Tedavi ve izlem
Tedavi, ketoasidoz veya hiperammonemi gibi yaşamı tehdit eden belirtiler çözüldüğünde ve karnitinle veya karnitinsiz (çoğunlukla cblA’da etkilidir) B12 vitamini kas içi enjeksiyonları yapılması gereken protein kısıtlı bir diyet içerir. Çoğu cblA hastasında ve neredeyse yarım cblB hastasında kobalamin takviyesine iyi bir yanıt bildirilmiştir. Oral antibiyotikler bağırsak florasından propionik asidi azaltmak için de yararlı olabilir.
Hastaların hayatın ilk yıllarında haftada bir değerlendirilmeli böbrek ve diğer organ fonksiyonları incelenmelidir. Dekompanzasyon önlenmeli ve protein alımı azaltılan hastaların diyetleri ailesiyle birlikte incelenmeli ve uygulanmalıdır. Hastalara acil durumlar için bileklik edinilebilir.
Ayrıca hastalığın seyrinde oluşabilecek sorunlara alakalı hasta fizyoterapist, psikiyatrist, çocuk nöroloğu ve gerekli diğer uzmanlar ile iş birliği içerisinde olmalıdır.
Prognoz
Prognoz, kompleman sistemine göre değişir, en uygun prognoza sahip cblA hastaları (30 yaşına kadar çoğu hasta iyi) ve cblB hastaları daha az elverişlidir. Hasta sayısı az olmasına rağmen, cblDv2, cblA’ya benzer görünür. Uzun süreli hayatta kalan hastalarda bir komplikasyon kronik böbrek yetmezliğidir.
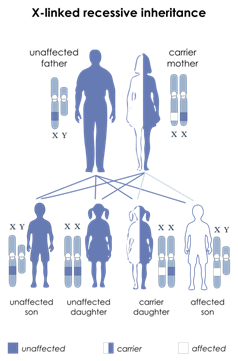
X-bağlı agammaglobulinema , primer bağışıklık
yetmezlik ile karakterize, immünoglobulinlerin(bağışıklık sistemi tarafından
üretilen özelliği enfeksiyonlarla savaşmaya yardımcı olmak olan proteinlerdir) çok düşük seviyelerde olması durumudur. Bu
durumdan etkilenen insanlarda genellikle yaklaşık 6 aylıktan itibaren sık ve
tekrarlayan bakteriyel enfeksiyonlar geliştirmeye başlar. Genel olarak teşhis
edilen enfeksiyonlar arasında akciğer enfeksiyonları (pnömoni ve bronşit), orta
kulak enfeksiyonları, konjonktivit , sinüs enfeksiyonları, çeşitli cilt
enfeksiyonları ve kronik diyare ile ilişkili enfeksiyonlar bulunur. X’e bağlı agammaglobulinemi, BTK’da geninde
oluşan değişikliklerden kaynaklanır (mutasyonlar) ve bir X’e bağlı resesif tavır
sergiler.
Klinik Tanım
Etkilenen bireyler, tortu şeklindeki materyal
immünoglobulinler nedeniyle yaşamın ilk birkaç ayında sağlıklıdır. Hastaların
çoğunda, yaşamın ilk iki yılında en sık S. pneumoniae ve H. influenzae’nin
neden olduğ, tekrarlayan veya kalıcı
bakteri enfeksiyonları gelişir: otitis media, konjonktivit, sinüzit, solunum
yolu enfeksiyonları, ishal ve cilt enfeksiyonları (impetigo, selülit , apseler
ve furuncles). Diğer ciddi enfeksiyonlar arasında ampiyem, menenjit, sepsis
veya septik artrit sayılabilir. Piyoderma veya selülit (nötropeni ile ilişkili)
ve psödomonas veya stafilokok sepsisi, özellikle 12 aylıktan küçük hastalarda
sık görülen bulgulardır. Lenf düğümleri, bademcikler ve diğer lenfoid dokular alışılmadık
derecede küçük veya yoktur. Nadir hastaların vitiligo, eritematöz döküntü, veya
alopesi totalis. Enfeksiyonlar yetişkinlik boyunca devam etme eğilimindedir.
Etkilenen hastaların şiddetli ve kronik enteroviral enfeksiyonlara karşı daha
yüksek duyarlılığa sahip oldukları bildirilmektedir. Büyüme ve gelişme
genellikle normaldir. Bazı hastalar daha az şiddetli klinik gösterime sahiptir
ve 10 yaşına kadar veya sonrasında immün yetmezlik olarak kabul edilmez. X’e
bağlı agammaglobulinemi (XLA) komplikasyonları arasında ilerleyici akciğer
hastalığı, kronik sinüzit, inflamatuar bağırsak hastalığı, artrit ve nörolojik
değişiklikler bulunur.
Semptomlar
Etkilenen bebekler genellikle tekrarlayan
bakteriyel enfeksiyonlar gelişene kadar yaşamın ilk birkaç ayında sağlıklıdır. En
yaygın bakteriyel enfeksiyonlar kulak enfeksiyonları , pnömoni , pembe göz ,
sinüs enfeksiyonları ve kronik ishale neden olan enfeksiyonlardır . Bu
bakteriyel enfeksiyonlar şiddetli ve hayatı tehdit edici olabilir. Etkilenen
kişilerin çoğu virüslerin neden olduğu enfeksiyonlara karşı savunmasız değildir.
Enfeksiyonlar genellikle uygun tedavi ile önlenebilir.
İnsanların% 80-99’u bu
semptomlara sahiptir; bademciklerde anormallik,kronik ishal ,kronik orta kulak
iltihabı ,konjonktivit (Pembe göz).
Epidemeyolojisi
Tahmini yaygınlık 1 / 350.000 ila 1 /
700.000’dir. Yıllık oran bilinmemektedir. Bozukluk dünya çapında çeşitli etnik
gruplarda bildirilmiştir. Sadece erkekler etkilenir ve kadınlar asemptomatik
taşıyıcılardır.
Etolojisi
X-e bağlı agammaglobulinemiye, B lenfosit
farklılaşması ve olgunlaşmasında rol oynayan BTK genindeki mutasyonlar neden
olur.
Teşhis
Genetik veya nadir bir hastalık için teşhis
koymak genellikle zor olabilir. Sağlık uzmanları, bir tanı koymak için
genellikle bir kişinin tıbbi geçmişine, semptomlarına, fiziksel muayenesine ve
laboratuvar test sonuçlarına bakar. Teşhis hakkında sorularınız varsa, bir
sağlık uzmanıyla görüşmelisiniz.
Yönetim ve Tedavi
XLA için iyileştirici bir tedavi yoktur, ancak
tutarlı gammaglobulin tedavisi ile iyi hastalık kontrolü sağlanabilir. Bu
tedavi, intravenöz bir şekilde (3 ila 4 haftada bir 400-600 mg / kg) veya deri
altından (haftada 100 mg / kg) verilebilir. Terapi mümkün olduğunca erken
başlatılmalıdır. Bazı immünologlar kronik profilaktik antibiyotikleri, akut
enfeksiyonların tedavisinin uzatılmasını ve maksimum dozda antibiyotik
kullanılmasını savunurlar.
Prognoz
XLA’lı hastaların çoğu normal bir yaşam
sürmektedir. Bazı hastalarda ciddi enfeksiyonlar ve kronik pulmoner hasar gibi
komplikasyonlar nedeniyle yaşam beklentisi azalabilir. Erken tedavi önlemleri
ve tedaviye uyum başlıca prognostik faktörlerdir
Homosistinüri
ile Metilmalonik Asidemi, vücudun amino asitler, lipidler ve
kolesterol dahil olmak üzere gıdalardan belirli besinleri düzgün bir şekilde işleyemediği
kalıtsal bir hastalıktır. Bu bozukluğa sahip olan insanlar iki ayrı durumdaki
özelliklerin kombinasyonuna sahiptir: “Metilmalonik asidemi” ve
“homosistinüri”. Hastalık yaşamın erken dönemlerinde başladığında bebekler kilo
almakta zorluk yaşarlar (gelişmede başarısızlık), beslenme güçlükleri çekerler
ve soluk bir görünüme sahip olurlar. Bebeklerde ayrıca zayıf kas tonusu
(hipotoni) ve nöbetler de gözlenebilir.
Bu duruma sahip
bebeklerin ve çocukların çoğu alışılmadık derecede küçük bir kafa büyüklüğüne
(mikrosefali), zihinsel engel ve gelişimsel gecikmeye sahiptir.
Durumun diğer özellikleri
arasında göz problemleri ve megaloblastik anemi adı verilen bir kan bozukluğu
bulunur. Ancak bu özellikler diğerlerine göre daha az yaygındır.
Bozukluk, ergenlik veya
yetişkinlikte başladığında semptomlar genellikle davranış ve kişilik
değişiklikleri, bilişsel sorunları (öğrenme, hafıza, algı,vb. ile ilgili
sorunlar) içerir. Ayrıca, bazı durumlarda yetenekler kaybolabilir. Bu da
performansın azalmasına, hafıza ve konuşma sorunları, uyuşukluk oluşmasına
neden olur.
Homosistinüri ile Metilmalonik Asidemi, birkaç
genden (MMACHC, MMADHC, LMBRD1, ABCD4 veya HCFC1)birinde oluşan
mutasyonlar nedeniyle de meydana gelebilir. Bu genlerdeki mutasyonlar sırasıyla “cblC, cblD, cblF, cblJ ve cblX ”
şeklindeki bozuklukların farklı türlerini açıklamaktadır. Bu koşullar için
tedavi kas içi hidroksikobalamin, oral betain ve folik asit enjeksiyonlarını
içermesinin yanında genel olarak herhangi bir tedavi bulunmamaktadır.
Belirtiler
Çoğu
hastalık için semptomlar kişiden kişiye göre değişmektedir. Aynı hastalığı olan
insanlar listelenen tüm semptomlara sahip olmayabilir ve bu bilgiler “İnsan
Fenotip Ontolojisi (HPO)” adı verilen
bir veritabanından gelir. HPO, tıbbi kaynaklarda açıklanan semptomlar hakkında
bilgi toplar.
Aşağıdaki
bilgiler bu hastalığa sahip bireylerde görülebilecek semptomlardır ve bu
hastalığa sahip insanların %80-%99 gibi bir kısmında bu semptomlar görülmektedir:
Tıbbi Terimler: Diğer Adları: Amblyopia Göz tembelliği
Gelişememe Gelişmede gecikme, kilo almada sorunlar
Yorgunluk
Yeme sıkıntıları
Teşhis
Herhangi bir hastalık hakkında teşhis koymak çoğu zaman zor
olabilir. Doktorlar teşhisi koymak için öncelikle hastanın tıbbi geçmişini,
hangi semptomlara sahip olduklarını öğrenirler, hastaya fiziksel testler
uygularlar ve yapılan testlerin sonuçlarını incelerler.
Test Kaynakları:Hastaya,
hastanın genetik bilgileri ile ilgili sonuçlar verecek testler uygulanır.
Yenidoğan Taraması: “Bebeğin İlk Testi”, ailelerin
yenidoğan taramasıyla ilgili eğitildiği bir eğitim merkezidir. Bu site,
taramalar hakkında bilgi ve kaynaklar sağlamaktadır.
“Ulusal Yenidoğan Taraması ve Küresel Kaynak Merkezi”,
sağlık profesyonelleri, halk sağlığı topluluğu, tüketiciler ve hükümet
yetkililerine fayda sağlamak için yenidoğan taraması ve genetik alanında bilgi
ve kaynak sağlamaktadır.
Tedavi
Aşağıda listelenen ilaçlar bu durumu tedavisi için “Gıda
ve İlaç Dairesi (FDA)” tarafından onaylanmıştır:
Betaine (Marka Adı: Cystadane): Yüksek homosistein kan
seviyelerini düşürmek için yapılan homosistinüri tedavisi.
Betaine:Osmoregülasyondaki rolü ile
ilgilenen doğal olarak oluşan bir bileşik. Bir ilaç olarak, betain hidroklorür
hipoklorhidri tedavisinde bir hidroklorik asit kaynağı olarak kullanılır.
Ayrıca, karaciğer bozukluklarının tedavisinde,
hiperkalemi, homosistinüri ve gastrointestinal rahatsızlıklar için de kullanılmaktadır.
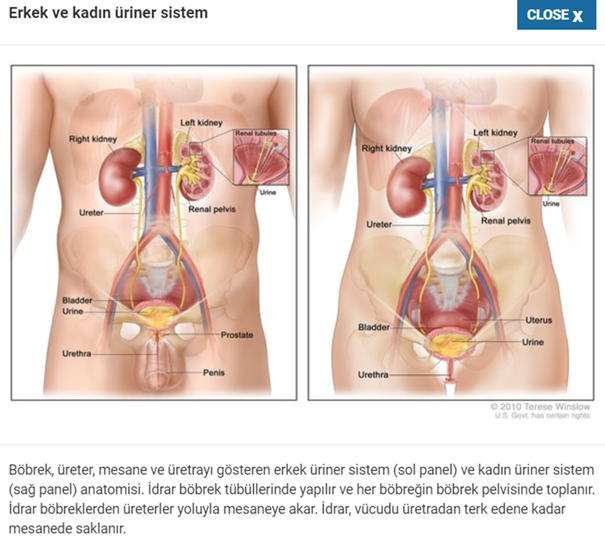
Fraiser
sendromu, psödohermafroditizm ve progresif glomerülopati ile tanımlanan nadir
bir hastalıktır.Hastalar normal kadın dış genital organları, çizgi gonadları ve
XY karyotipi ile başvurur ve sıklıkla gonadoblastom gelişir. Glomerüler
semptomlar, ergenlik veya erken yetişkinlikte son dönem böbrek
yetmezliğine ilerleyen spesifik olmayan fokal ve segmental glomerüler skleroz
ile karakterize çocukluk çağı proteinüri ve nefrotik sendromdan oluşur.
Frasier
sendromu böbrekleri ve genital organları etkileyen bir
durumdur. Etkilenen bireyler, bazı glomerüllerde skar dokusunun oluştuğu,
böbreklerdeki kanı atıktan süzen
küçük kan damarları olan skar dokusunun oluştuğu fokal segmental
glomerüloskleroz adı verilen bir duruma sahiptir.Frasier sendromlu kişilerde ,bu
durum genellikle ergenlik döneminde böbrek yetmezliğine yol açar.
Frasier
sendromlu erkeklerde tipik erkek kromozom paterni olmasına
rağmen ( 46, XY),
dış genital organların açıkça erkek veya açıkça kadın (belirsiz genital) veya
genital organların tamamen dişi göründüğü gonadal disgenezisi vardır. İç
üreme organları (gonadlar) tipik olarak gelişmemiş ve çizgi gonadları olarak
adlandırılmıştır. Bu anormal gonadlar işlevsizdir ve genellikle kanserli
hale gelir, bu nedenle genellikle ameliyatın erken dönemlerinde çıkarılırlar.
Etkilenen dişilerin genellikle normal cinsel organları ve gonadları vardır ve durumun sadece böbrek özelliklerine sahip. Durumun tüm özelliklerine sahip olmadıkları için, kadınlara genellikle izole nefrotik sendrom tanısı verilir.
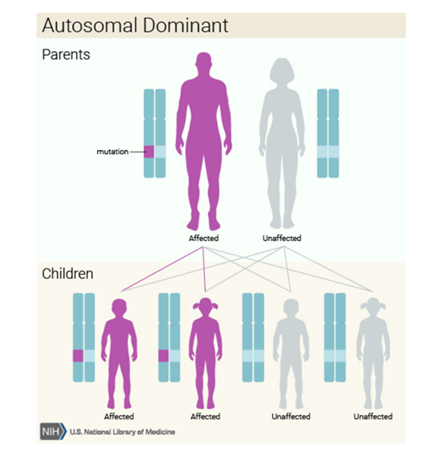
Kalıtım Kalıbı
Bu durum otozomal dominant bir paternde kalıtsaldıryani her hücredeki değiştirilmiş genin bir kopyası bozukluğa neden olmak için yeterlidir.
Klinik Tanım
Moorthy ve diğ. (1987) , Denys-Drash sendromu vakaları olarak bildirilen bazı hastaların aslında Fraiser sendromu adını önerdikleri farklı bir bozukluğa sahip olduklarını ileri sürmüşlerdir. Moorthy ve diğ. (1987) , daha önce bildirilen 6 çizgi gonad, psödohermafroditizm ve böbrek yetmezliği olan hastayı tartışmıştır. Bazı hastalarda tanı, primer amenore değerlendirmesi sırasında başarılı bir böbrek nakli sonrasında konmuştur. 6 hastanın 5’inde çizgi gonaddan kaynaklanan gonadoblastom saptandı.
Fraiser
sendromu ile ilişkili 3 hasta tanımlamışt. Her üçü de 2-6 yaş arasında
kalıcı proteinüri ile başvurdu ve daha sonra 9-35 yaşları arasında son
dönem böbrek yetmezliğine ilerleyen nefrotik sendrom gelişti. Böbrek
yetmezliğinin başlamasından önce yapılan böbrek biyopsilerinde 1 hastada
minimal nonspesifik glomerüler değişiklikler, diğer 2 hastada fokal ve
segmental glomerüler skleroz saptandı. Her üç hastaya da nefrotik sendrom nüksü
olmadan başarılı böbrek nakli yapıldı. Normal kadın fenotipi olan bu 3
kadında primer amenore değerlendirmesi 46, XY gonadal disgenezis tanısına yol
açmıştır. Üç hastadan birinde 19 yaşında teşhis edilen gonadoblastom
gelişti; cerrahi tedaviden sonra nüks gözlenmedi. Diğer 2 hastaya
bilateral cerrahi gonadektomi yapıldı.
Melo ve diğ. (2002) , Denys-Drash sendromunun dış genital özelliğine sahip Fraiser sendromu tanısı alan bir hastayı bildirmiştir. Bu 2 sendromunun farklı hastalıklar olmadığını, ancak WT1 genindeki değişikliklerin neden olduğu bir spektrumun 2 ucunu temsil edebileceğini öne sürdüler.
Görülme Sıklığı
Frasier sendromunun nadir bir durum olduğu düşünülmektedir; bilimsel literatürde yaklaşık 50 vaka tanımlanmıştır.
Moleküler
Genetik
WT1 geninin
ekson 8 veya 9’a mutasyonlar Denys-Drash 10 ilgisiz hastalarda tespit edilmiş
olduğundan sendromu ile Pelletier et al.(1991) ,
Fraiser sendromlu hastalarda WT1’in 1 ila 10 eksonlarını taradılar ,
ancak hiçbir mutasyon tespit etmediler. Tek iplikli konformasyon
polimorfizm (SSCP) analizi ile mutasyon taraması yaptıkları için, nokta mutasyonlarının
sadece tahmini %80’ini tespit eden bir yöntem olduğundan, genin başka
yerlerinde mutasyon olasılığı tamamen dışlanmamıştır.
Berta ve diğ. (1992) ,
XY gonadal disgenezisi ve kronik böbrek yetmezliği olan 2 kız çocuğunda Y kromozomunun
cinsiyet belirleyici bölgesinde veya SRY genindeki mutasyonlarda ( 480000 ) büyük bir delesyon
bulamadılar .
3 hastada
Fraiser sendromu, Barbaux ve diğ. (1997) , WT1 geninin intron 9’unun ( 607102.0018 ; 607102.0019 ) donör
ekleme alanındaki mutasyonları, + KTS izoformu olarak bilinen bir kayıp
ile tanımladılar . Normal
olarak, intron 9’daki alternatif bir ekleme yeri, WT1 proteininin üçüncü ve
dördüncü çinko parmakları arasına 3 amino asit (KTS) eklenmesine izin
verir. 3 hastanın hepsinde erkek psödohermafroditizmi, nefrotik sendrom
vardıson dönem böbrek yetmezliğine ilerleyen ve 46, XY gonadal
disgenezi. Böbrek yetmezliğinin başlamasından önce yapılan böbrek
biyopsilerinde 1 hastada minimal nonspesifik glomerüler değişiklikler, diğer 2
hastada fokal ve segmental glomerüler skleroz saptandı. Bir hastada 19
yaşında gonadoblastom teşhis edildi.
Klamt ve diğ. (1998) ,
Fraiser sendromuna neden olan WT1 mutasyonları tarafından hiçbir mutant
proteinin üretilmediğini göstermiştir . Bunun yerine, mutasyon,
proteinin 2 ilave izoformunun, fazladan 3 amino aside (KTS) sahip olan ve
olmayanlara değiştirilmiş bir oranla sonuçlanır. Denys-Drash sendromunda,
tümör riski Fraiser sendromundan çok daha fazladır . Baskın
negatif mutant aleli arızalıdır ve ikinci alelin kaybı (2 vuruşlu modele göre)
tümör oluşumunda önemli bir adım olabilir. Aksine, Fraiser hastaların
sadece daha kısa bir izoform üretebilen 1 normal WT1 ve 1 kopyası
vardır. Bu nedenle alel kaybı, WT1’in + KTS izoformunu üretemeyen, ancak
yine de büyük miktarlarda -KTS izoformuna sahip olan hücrelere yol
açacaktır. Bu bakımdan, çıplak farelerde G401 Wilms tümör hücre dizisinin
tümör oluşumunun + KTS ve -KTS izoformları ile aynı ölçüde bastırılabileceğini
belirtmek ilginçtir. Gonadoblastoma sık olduğu Frasier hastalar.
Melo ve diğ. (2002) ,
bağlanma yeri kullanımında bir değişiklik öngören IVS9 + 4C-T mutasyonu
( 607102.0018 ) olan
Fraiser sendromlu 19 yaşında bir erkek
olduğunu bildirmiştir . Sıradışı bir fenotipi vardı. WT1
transkript analizi FS tanısını doğrulayan normal pozitif / negatif KTS izoform
oranının tersine döndüğünü gösterdi. Yazarlar, bu hastanın Denys-Drash
sendromun dış genital özelliğine sahip olduğu ve bu 2 sendromun ayrı
hastalıklar olmadığını, ancak WT1 genindeki değişikliklerin neden olduğu bir
spektrumun 2 ucunu temsil edebileceği sonucuna vardı .
Nedenler
WT1 genindeki mutasyonlar Frasier
sendromuna neden olur . WT1 geninin spesifik
bölgelerine bağlanması (bağlanma) ile, diğer genlerin aktivitesini düzenleyen
bir protein yapmak için yönergeler DNA. Bu
eyleme dayanarak, WT1 proteinine transkripsiyon faktörü denir. WT1
proteini böbreklerin
ve gonadların gelişiminde
rol oynar (kadınlarda yumurtalıklar ve erkeklerde testisler) doğumdan
önce.
Frasier
sendromuna neden olan WT1 gen mutasyonları, gen
aktivitesini kontrol etme ve böbreklerin ve üreme organlarının gelişimini
düzenleme yeteneğine sahip bir proteinin üretilmesine
yol açarak Frasier sendromunun belirti ve
semptomlarına neden olur.
Frasier sendromu, WT1 genindeki mutasyonların da neden olduğu Denys-Drash sendromu adı verilen başka bir duruma benzer özelliklere sahiptir . Bu iki durum genetik bir nedeni paylaştığı ve üst üste binen özelliklere sahip olduğu için, bazı araştırmacılar bunların iki farklı koşulun değil, bir spektrumun parçası olduğunu ileri sürmüşlerdir.
Normal İşlev
WT1 geni doğumdan önce (erkeklerde kadınlarda yumurtalıkların ve testislerin) böbrekler ve üreme organlarına gelişimi için gerekli olan bir proteini yapmak için talimatlar sağlar. Doğumdan sonra, WT1 protein aktivitesi glomerulus olarak bilinen ve böbrekler yoluyla kanı süzen bir yapı ile sınırlıdır. WT1 proteini, hücre büyümesinde, hücrelerin belirli işlevleri yerine getirmek için olgunlaştıkları (farklılaşma) ve hücrelerin kendi kendini yok etmelerinde (apoptoz) rol oynar. Bu işlevleri yerine getirmek için WT1 proteini, DNA’nın belirli bölgelerine bağlanarak (bağlanarak) diğer genlerin aktivitesini düzenler. Bu eyleme dayanarak, WT1 proteinine transkripsiyon faktörü denir.
Etolojisi
Frasier
sendromu, WT1 geninde (11p13) intron 9’un (önceden IVS9 +
4; IVS9 + 5 olarak anılacaktır) 4-5 nükleotidlerini etkileyen spesifik
patojenik varyantlarla ilişkilendirilmiştir. WT1, hem böbrek hem de
gonadal gelişim için önemli olan düzenleyici transkripsiyon faktörü olarak
işlev gören bir proteini kodlar.
Hastalık Belirtileri
Bu tablo, bu hastalığı olan kişilerin sahip olabileceği belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye değişir. Aynı hastalığı olan insanlar listelenen tüm semptomlara sahip olmayabilir. Bu bilgiler İnsan Fenotip Ontolojis (HPO) adı verilen bir veritabanından gelir.. HPO, tıbbi kaynaklarda açıklanan semptomlar hakkında bilgi toplar. HPO düzenli olarak güncellenir. Bir belirti hakkında daha ayrıntılı bilgiye erişmek için HPO Kimliğini kullanın
Teşhis Yöntemleri
Histolojik
analizde FSGS bulguları ile ilerleyen glomerulopatinin çocukluk başlangıcında
tanıdan şüphelenilmektedir. Ergenlik gecikmesi veya primer amenore olan
fenotipik dişiler nefropati belirtileri açısından dikkatle
değerlendirilmelidir. Klinik bulgular WT1 ile ilişkili
bozuklukların teşhisini önerdiğinde, sıcak noktanın 8-9 tek gen
testieksonbitişik intronlar ile gerçekleştirilebilir. Karyotip
testi, WT1 intron 9 patojenik varyantları olan tüm
bireyler için önerilir.
Genetik veya
nadir bir hastalık için teşhis koymak genellikle zor olabilir. Sağlık
uzmanları, bir tanı koymak için genellikle bir kişinin tıbbi geçmişine,
semptomlarına, fiziksel muayenesine ve laboratuvar test sonuçlarına
bakar. Aşağıdaki kaynaklar, bu durumun teşhisi ve testi ile ilgili bilgi
sağlar. Teşhis hakkında sorularınız varsa, bir sağlık uzmanıyla
görüşmelisiniz.
Yöntem ve Tedavi
Yönetim çok
disiplinlidir ve aşağıdakileri içermelidir: nefrolog kronik böbrek
yetmezliğinin tedavisi için (başlangıçta nefroprotektif tıbbi tedavi ile ve
daha sonra ESRD oluştuğunda böbrek replasman tedavileri veya transplantasyon
ile), endokrinologlar ilişkili testis gelişimi bozukluğunun tedavisi
için ve onkologlarve cerrahlar tümör oluşumunu önlemek için erken bir
gonadektomi ihtiyacını değerlendireceklerdir. Böbrek nakli veya peritoneal
yerleşim sırasında preemptif bilateral gonadektomidiyaliz sonda bir
seçenek olabilir.
Prognoz
Yaşam
beklentisi hakkında sınırlı bilgi vardır. Böbrek nakli sonrası nefrotik
sendrom tekrarlamaz. 46, tam gonadal disgenezi olan XY bireyleri kısırdır.
Yaşam beklentisi hakkında sınırlı bilgi vardır. Böbrek nakli sonrası
nefrotik sendrom tekrarlamaz. 46, tam gonadal disgenezi olan XY bireyleri
kısırdır.
Sakati sendromu
“Akrosefalopolisindaktili” (ACPS) olarak bilinen nadir bir genetik grubuna ait
çok nadir bir hastalıktır. ACPS’nin tüm türleri kafatasındaki bazı kemiklerin
arasındaki lifli eklemlerin erken kapanması sonucu kafanın üst kısmının sivri
olmasına, bazı el ve ayak parmakları arasında bağsı dokusu oluşuna veya
birleşmelere ve/veya normal sayıdan daha fazla el, ayak parmağına sahip olmakla
kategorize edilir.
Ek olarak, ACPS tip III
olarak bilinen Sakati sendromu, ayak kemiklerindeki anormalliklere, doğumda
olan yapısal kalp kusurlarına ve diğer bulgulara neden olur.
Sakati sendromu
bilinmeyen sebeplerle rastgele oluşan yeni bir genetik değişim (mutasyonlar) nedeniyle
oluştuğu düşünülüyor. Bu mutasyon otozomal baskın olarak kalıtılır.
Belirti
ve Semptomlar
Sakati sendromunda, kafatasındaki
kemikler arasındaki lifli eklemler erkenden kapanmasıyla bebeklerin kafasının
yukarıya doğru hızlı büyümesine neden olur. Bunun sonucunda, kafaları uzun, dar
ve sivri oluşur.
Etkilenen bireyler ayrıca düz,
anormal küçük bir yüz; çıkıntılı gözler; gözler arasında anormal geniş bir
boşluk; uzamış bir burun; geniş, hatalı biçimlendirilmiş ve düşük pozisyonda kulaklar;
ve öne çıkan bir alın gibi alışılagelmeyen yüz karakterlerini içerir.
Sakati sendromu eller ve ayakları
içeren anormal kısa parmaklar, alışılmadık geniş başparmaklara ve büyük ayak
parmaklar, yapışık ve perdeli ayak parmaklar ve normal sayıdan daha fazla el ve/veya
ayak parmak oluşumlarını içeren ciddi bozukluklarla karaterize edilir.
Bacaklardaki anormallikler eğri
dar kemikler olmak üzere, anormal yapılı yer değiştirmiş baldır kemikleri ve az
gelişmiş kaval kemiklerini de içerir. Ayrıca, hem ayaklarda hem de kollar
normale göre daha kısadır.
Bu bozuklukla birlikte ilave olan
semptomlar; birlikte sıkışmış dişler, az gelişmiş bir üst çene kemiği, öne
doğru çıkmış çene kemiği, kısa bir boyun, düşük saç çizgisi, saç eksikliği ve doğuştan
kalp hastalığı içerebilir.
Nedenleri
Sakati sendromunun tam olarak sebebi anlaşılamamıştır. Yeni ya da aralıklı,
baskın genetik değişimler olabileceği düşünülüyor. Böyle bir mutasyonun asıl
sebebi açığa çıkmamasına rağmen, bazı araştırmacılar anne ve babanın ilerlemiş
yaşının bu durumun oluşmasında etkisinin olabileceğini düşünüyorlar.
Sakati sendromlu bir kişi çocuk sahibi olsaydı, hastalığın değişmiş geni
otozomal baskın bir özellik olarak geçebilir. Genetik hastalıklar biri babadan
diğeri anneden gelen iki baskın genle belirlenir.
Baskın genetik hastalıklar,
anormal kopyalı genin sadece bir kopyasının bile yeterli olduğu durumlarda
oluşur. Bu anormal gen ebeveynlerden kalıtılabilir veya etkilenen bireyde yeni
bir mutasyon sonucu oluşabilir. Anormal genin, etkilenen ebeveynlerden
çocuklarına geçme riski çocuğun cinsiyetine bakılmaksızın her bir hamilelik
için %50’dir.
Benzer Hastalıklar
Aşağıda bahsedilen bozukların belirtileri Sakati sendromu ile benzer
olabilir.Karşılaştırmalar ayırıcı tanı için yararlı olabilir.
Akrosefalopolisindaktili (ACPS); Noack sendromu (Tip I), Carpenter sendromu
(Tip II), Sakati sendromu (Tip III) ve Goodman sendromu (Tip IV) gibi çok nadir
genetik bozuklukların bir grubudur. Bütün bu bozukluklar normalden daha fazla
el ve/veya ayak parmakları ve bitişik el ve/veya ayak parmakları ve sivri
görünen uzun, dar bir kafa ile karakterize edilir.
Carpenter sendromu ellerde anormal kısa yapışık parmaklar ve beşten fazla,
yapışık da olabilen, ayak parmakları; uzun, dar bir kafa ile karakterize olan
çok nadir bir kalıtsal bozukluktur. Etkilenen bireyler aşağı doğru eğilmiş
gözlere, düzleştirilmiş bir burun köprüsüne, geniş yanaklara, düşük pozisyonda
kulaklara ve az gelişmiş çene kemiği karakterine sahiptir. Diğer özellikler obezite
başlangıcı, zeka geriliği, bağırsak duvarı boyunca bağırsak kısımlarının
çıkıntısı, az gelişmiş cinsel organlar, ve doğumsal kalp hastalığıdır.
Carpenter sendromu otozomal resesif bir özellik olarak kalıtılır.
Goodman sendromu, uzun, dar bir kafa; birçok yüz kusuru; perdeli parmaklar
ve/veya ayak parmakları; ekstra parmaklar ve anaormal şekilde eğimiş ve kalıcı
şekilde bükülmüş beşinci parmaklarla karakterize edilmiş çok nadir görülen bir
genetik hastalıktır. Diğer özellikler önkol kemiklerinden birinin ayrılması,
dizlerin anormal yakın ve anormal uzak ayak bilekleri ve doğuştan kalp
rahatsızlıklarının bulunmasıdır. Zeka normal sınırlar içindedir. Bazı
araştırmacılar Goodman sendromunun Carpenter sendromunun başka bir türü
olduğunu düşünmektedir.Goodman sendromu otozomal resesif bir özellik olarak
kalıtılır.
Apert sendromu perde-şekilli ve sivri bir kafa; öne çıkan bir alın; ortası
düz görünen bir yüz; ve dışarı çıkmış, şaşı gibi görünen ve/veya geniş ayrılmış
gözler olarak karakterize edilen nadir bir genetik bozukluktur. El ve ayaklarda
perdeli ve/veya birleşmiş el ve ayak parmakları ve genellikle ikinci ila dördüncü
parmaklarda tek bir tırnak gibi anormallikleri içerebilir. Diğer özellikleri sıkışık üst dişler, çıkık bir
çene, alışılmadık yüksek ve sivri bir çene, düşük pozisyonda kulaklar, işitme
kaybı ve zeka geriliği içerebilir. Apert sendromu otozomal baskın bir özellik
olarak kalıtılır.
Teşhis
Sakati sendromu, klinik
değerlendirme ve fiziksel bulguların saptanmasına dayanarak doğumda tespit
edilebilir.
Tedavisi
Tedavi öncelikle kusurlu
oluşumların cerrahi yöntemle düzeltilmesini içerir. Erken kranyofasiyel
ameliyat, kafatasındaki kemiklerin erken kapanmasını düzeltmek için
uygulanabilir ve ek olan kranyofasiyel ameliyat, ayaklar ve ellerdeki
anormallikleri düzeltmek için yapılabilir. Ayrıyeten, ayak kemiklerinmdeki
anormalliklerin cerrahi düzeltilmesi bireylerin yürümesini de geliştirebilir.
Doğuştan kalp rahatsızlığı olan
Sakati sendromlu bebekler de cerrahi olarak tedavi edilebilir. Uygulanan
cerrahi prosedür, kalp rahatsızlığının ciddiyetine, yerine ve bunlara bağlı semptomlara bağlı olacaktır.
Diğer tedavi semptomik ve
destekleyicidir. Genetik danışmanlık etkilenen bireyler ve aileleri için
faydalı olacaktır.
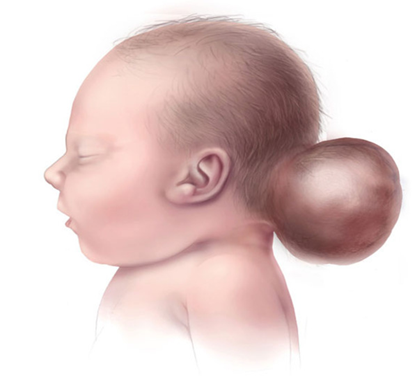
Meckel sendromu, böbreklerde çoklu kistler, kafatasındaki bir açıklıktan (oksipital ensefalosel) beynin bir kısmının çıkıntısı ve ekstra parmaklar (polidaktili) ile karakterize çok ciddi bir hastalıktır. Etkilenen çocuklarda baş ve yüz, karaciğer, akciğerler, cinsel organlar ve idrar yollarını etkileyen anormallikler de olabilir. Bu ciddi sağlık sorunları nedeniyle, Meckel sendromlu bebeklerin çoğu doğumdan sonra uzun süre hayatta kalmaz. Meckel sendromu, sekiz genden birinde mutasyonlardan kaynaklanır ve otozomal-resesif bir şekilde kalıtsaldır.
Belirti ve
Semptomlar
Meckel sendromu ile ilişkili spesifik semptomlar bir kişiden
diğerine büyük ölçüde değişir. Etkilenen çocuklar aşağıda ayrıntılı
semptomların hepsine sahip olmayacaktır. Merkezi sinir sistemi, pulmoner veya
böbrek anormallikleri her zaman perinatal ölümle sonuçlanır.
Meckel sendromuyla ilişkili en yaygın merkezi sinir sistemi anormalliği, bir bebeğin kafatasında bir boşlukla doğduğu bir durum olan oksipital ensefaloseldir.
Etkilenen bebekler anormal derecede küçük bir çene (mikrognati),
genişlemiş ve kusurlu kulaklar, yarık damak, yarık dudak, eğimli alın ve kısa
boyun gibi farklı özelliklere sahip olabilirler. Etkilenen çocuklarda anormal
derecede küçük gözler (mikroftalmi) ve gözlerin sinirlerinin az gelişmesi
(optik sinir hipoplazisi veya koloboma) dahil olmak üzere göz (oküler)
anormallikleri görülebilir. Böbreklerdeki çoklu kistler (multikistik böbrek
displazisi) Meckel sendromuyla ilişkili en yaygın semptomdur.
Etkilenen bireyler ayrıca ekstra parmak ve ayak parmaklarına, çoğunlukla ellerin baş parmağında ekstra parmaklara (postaksiyal polidaktili) sahip olabilirler.
Genetik
Görülme Sıklığı
Yaygınlık Avrupa’da 50,000’de 1 doğum olarak tahmin edilmektedir. Canlı
doğum yaygınlığının dünya çapında 13,250’de 1 ile 140,000’de 1 arasında olduğu
bildirilmektedir. Canlı doğum yaygınlığı, Fin popülasyonunda (1/9,000), Belçika
ve Kuveyt Bedevi popülasyonlarında (1/3,500) ve Gujarati Kızılderililerinde
(1/1,300) önemli ölçüde daha yüksektir. Cinsiyet veya etnik tercih
bildirilmemiştir.
Kalıtım
Paterni
Bu durum otozomal resesif bir desende kalıtsaldır, bu da her
hücredeki genin her iki kopyasının mutasyonları olduğu anlamına gelir. Otozomal
resesif bir duruma sahip bir bireyin ebeveynleri her biri mutasyona uğramış
genin bir kopyasını taşır, ancak tipik olarak durumun belirti ve semptomlarını
göstermezler.
Nüks riski %25’dir.
Nedenleri
Meckel sendromu, on üç gendeki değişikliklerden (mutasyonlar)
kaynaklanabilir: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRİP1L, TCTN2, TCTN3,
TMEM67, TMEM107, TMEM216, tmem231 ve TMEM237. Bu 13 gendeki mutasyonlar tüm
vakaların yüzde 75’ini oluşturur; kalan yüzde 25’in bilinmeyen genetik
nedenleri vardır. Bu genlerin çoğu, Joubert sendromu adı verilen nörolojik bir
bozukluktan da sorumludur ve Meckel sendromunun Joubert sendromunun aşırı
öldürücü formu olduğu kavramına yol açar.
Teşhis
Yöntemleri ve Tedaviler
Oksipital ensefalosel ve displastik böbrekleri gösteren fetal
ultrasonografi ile tanı konabilir. MKS tanısı için üç ana malformasyondan ikisi
veya bir klasik özellik ile birlikte diğer iki anomali yeterlidir. Otopsi de
gerekebilir. Bozukluk genellikle 14. gebelik haftasından önce tespit edilir.
Moleküler genetik testler, genetik danışmanlığa rehberlik etmek için tanıyı
doğrulamak için kullanılabilir.
Ölümcül bir sonucu olan Meckel sendromu için şu anda tedavi mevcut
değildir.
Jeune sendromu öncelikle kemikleri etkileyen nadir bir durumdur. Yaygın belirti ve semptomlar arasında akciğerlerin büyümesini ve genişlemesini kısıtlayan küçük bir göğüs ve kısa kaburgalar bulunur ve bu da genellikle hayatı tehdit eden solunum güçlüklerine neden olur. Diğer semptomlar kollarda ve bacaklarda alışılmadık şekilli kısaltılmış kemikleri içerebilir. Kemikler ve ekstra parmaklar veya ayak parmakları. Bebeklik döneminin nefes alma zorluklarından kurtulan insanlar daha sonra ciddi böbrek veya kalp problemleri geliştirebilir. Birçok durumda Jeune sendromunun nedeni bilinmemektedir; ancak, değişiklikler (mutasyonlar) birkaç farklı genler bu durumdakibazı ailelerde tanımlanmıştır. Jeune sendromumiras bir otozomal resesiftavır. Tedavi, her insanda mevcut olan belirti ve semptomlara dayanır.
Boğucu torasik distrofi olarak da adlandırılan Jeune sendromu, dar bir
toraks, kısa uzuvlar ve asetabula’nın ‘trident’ yönü ve metafiz
değişikliklerini içeren radyolojik iskelet anormallikleri ile karakterize kısa
bir kaburga displazisidir. Yıllık doğum sıklığı bilinmemektedir ancak 1-5 /
500.000 olduğu tahmin edilmektedir.
Polidaktili olan veya olmayan kısa kaburga torasik displazi (SRTD), dar bir torasik kafes, kısa kaburgalar, kısaltılmış tübüler kemikler ve asetabular çatının ‘trident’ görünümü ile karakterize edilen bir grup otozomal resesif iskelet sililpatisine işaret eder. SRTD, Ellis-van Creveld sendromunu (EVC) ve daha önce Jeune sendrom veya boğucu torakal distrofi (ATD), kısa kaburga-polidaktili sendromu (SRPS) ve Mainzer-Saldino sendromu olarak tanımlanan bozuklukları kapsar.
Kromozomal Konum
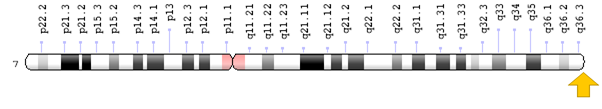
Sitogenetik Yer:
7q36.3, 36.3 konumundaki kromozom 7’nin uzun (q) kolu
Moleküler Yer: Kromozom 7’de baz çiftleri 158.839.245 ila 158.958.695 (Homo sapiens Güncellenmiş Ek Açıklama Bülteni 109.20191205, GRCh38.p13)
Bu Genin Diğer İsimleri
CFAP163
DIC6
FAP163
SRPS6
SRTD8
Jeune Sendromu’nun Eş anlamları
boğucu
torasik displazi
ATD
torasik-pelvik-falangeal
distrofi
boğucu
torasik kondrodistrofi
kondroektodermal
displazi benzeri sendrom
infantil
torasik distrofi
Jeune
torasik distrofisi
Etkilenen Popülasyonlar
ATD insidansı 100.000 ila 200.000
canlı doğumda yaklaşık 1’dir. Erkekler ve dişiler, çeşitli etnik veya
ırksal geçmişlere sahip kişiler gibi eşit sayıda etkilenmiş gibi görünmektedir.
Klinik Özellikler
Maroteaux ve Savart (1964) torasik distrofiyi boğucu olarak tanımladılar ve
göğüs kafesi, pelvis ve uzuvlardaki iskelet değişikliklerinin Ellis-van Creveld
sendromunda (EVC; 225500 ) gözlemlenenlere
benzer olduğunu belirttiler . Pirnar ve Neuhauser (1966) etkilenen 3
erkek kardeş bildirmiş ve tırnaklarının displazisi olmadan polidaktili
varlığını belirtmiştir. Erken çocukluk döneminde hayatta kalanlar, kronik
nefrit ( Wahlers, 1966 ) ve bağırsak malabsorpsiyonu
( Karjoo ve diğerleri, 1973 ) gibi
başka bozukluklar geliştirme eğilimindeydi .
Langer
(1968) , polidaktili olgularda, sadece radyolojik
nedenlerle Ellis-van Creveld sendromundan farklılaşmanın mümkün
olmayabileceğine dikkat çekmiştir . Polidaktili, ATD’nin
tutarsız bir özelliğidir ve mevcut olduğunda genellikle ayakları da
etkiler. Aksine, ellerin polidaktili EVC’de sabit bir özelliktir, ancak
ayaklar nadiren etkilenir. ATD’deki ana viseral anormallik renaldir, oysa
EVC’de kardiyaktır.
Shokeir
(1970) , Norveç ekstraksiyonunun boğucu torasik distrofisi ile
ilişkili 5 etkilenmiş kişiyi tanımlamıştır. Kistik böbrek değişiklikleri
(Potter tip IV) tanımlandı. Kistik lezyonlar böbrek, karaciğer ve
pankreasta ortaya çıkabilir ( Hopper
ve ark., 1979 ; Landing
ve ark., 1980 ).
Finegold
ve diğ. (1971) hipoplastik akciğerli bir olgu ve otopside alveol
sayısında belirgin bir azalma olduğunu bildirmişlerdir.
Oberklaid
ve diğ. (1977) 10 vaka bildirmiştir. Böbrek ve karaciğer
değişiklikleri progresifti ve en az 2 hastada ölüm nedeni böbrek yetmezliği
idi. Dikkate değer bir vaka, hala 15 yaşında ve boy için 25. yüzdelikte
yaşayan bir çocuğun vakasıydı. Küçük bir göğsü vardı, ancak tek radyolojik
bulgu kısa kaburgalardı. 32 yaşında bir hasta Friedman
ve ark. (1975) .
Turkel
ve diğ. (1985) otopside 7 yenidoğan vakasını incelemiş; 2’si
akraba ebeveynlerinden doğan kardeşlerdi. Cüce telaffuz
edilmedi; polidaktili de olan sadece bir bebekte uzuvlar
kısaydı. Enkondral ossifikasyon femur, omur ve kaburga bölümlerinde
düzensizdi. Pulmoner hipoplazi küçük toraks ile ilişkili
idi. Periportal fibroz, safra kanalı proliferasyonu, siroz (1 vakada) ve
değişken pankreatik fibroz da tarif edildi.
Whitley
ve diğ. (1987) , yenidoğan döneminde direkt hiperbilirubinemi ve
hepatik fibroz ile ilişkili karaciğer fonksiyon bozukluğunu tarif
etmişlerdir. Hudgins
ve diğ. (1990) , sirozla ilişkili progresif hepatik disfonksiyonu
olan bu bozukluğa sahip 2 sib tanımlamıştır. Giorgi
ve diğ. (1990) , hafif bir sendrom formuna sahip 2 kız
kardeş tanımlamıştır.
Zack
ve Beighton (1995) akraba çiftli karışık soydan 6
çocuğun 1’inde spondiloenfondromatoz
(bkz. 607944 ) adını
verdiklerini açıkladılar . İlk
olarak 2.5 yaşında görüldüğünde, psödoakondroplazi ( 177170 ) için geçici bir
tanı konulmuştur, ancak daha sonra özellikler spondiloenfondromatozun
radyolojik görünüm tanısına dönüşmüştür. Gez ile, pelvisin 2.5
yaşında konfigürasyonu, boğucu toraks displazisini bir şekilde
düşündürdü. Daha sonra, 13 yaşında çekilen fotoğraflarla belirtildiği
gibi, göğsün belirgin daralması gelişti.
Labrune
ve diğ. (1999) Jeune sendrom ve karaciğer hastalığı
klinik ve laboratuvar bulguları olan 3 çocuk
bildirmişlerdir . Karaciğer tutulumu şiddetliydi ve hepatik fibrozise
ve daha sonra portal hipertansiyonlu biliyer siroza yol açtı. Bir hastada
uzamış neonatal kolestaz ilk tezahürken, diğer 2 hastada fibroz ve hatta siroz
geliştiğinde hepatik lezyonlar geç fark edildi. Ursodeoksikolik asit ile
tedavinin, klinik ve laboratuvar verilerindeki iyileşmeye bağlı olarak hepatik
disfonksiyonun ilerlemesini kontrol ettiği görülmüştür. Yazarlar ,
serum safra asidi konsantrasyonu ölçümleri de dahil olmak üzere Jeune sendromlu hastalarda hepatik fonksiyonun
düzenli olarak takip edilmesi gerektiğini önerdiler .
Kajantie
ve diğ. (2001) , yenidoğan semptomları hafif solunum
sıkıntısından asfiksi ve ölüme kadar değişen ATD’li 3 sib tarif
etmişlerdir. Yazarlar, üçüncü trimesterden önce genç siblerin doğum öncesi
tanısında zorluklar olduğunu bildirmişlerdir. Şiddetli etkilenen
hastaların bile yeni yenidoğan yoğun bakım tedavi
seçenekleri göz önüne alındığında uygun bir prognoza sahip olabileceğini
önerdiler.
Normal İşlevler
WDR60 geni , WD tekrar protein ailesinin bir üyesini
kodlar. WD tekrarları, tipik olarak gl-his ve trp-asp (GH-WD) tarafından
desteklenmiş yaklaşık 40 amino asidin minimal olarak korunmuş bölgeleridir ve
heterotrimerik veya multiprotein komplekslerinin oluşumunu kolaylaştırabilir. Bu
ailenin üyeleri hücre döngüsü ilerlemesi, sinyal iletimi, apoptoz ve gen
regülasyonu dahil olmak üzere çeşitli hücresel süreçlerde yer
alır. Kodlanmış protein dört WD tekrarı içerir ve silya oluşumunda rol
oynayabilir. Bu gendeki mutasyonlar kısa kaburga polidaktili ve Jeune
sendromları ile ilişkilendirilmiştir.
Genetik Değişikliklere İlişkin Sağlık
Koşulları
Polidaktili olan veya
olmayan kısa kaburga torasik displazi 8 (SRTD8): Kısa kaburga torasik displazi,
bir grup otozomal resesif ciliopati ve daraltılmış torasik kafes, kısa tırtıklı
kemikler ile karakterize asetabular çatının görünümü. Polidaktili değişken
olarak mevcuttur. İskelet dışı tutulum, yarık dudak / damak yanı sıra beyin,
göz, kalp, böbrekler, karaciğer, pankreas, bağırsaklar ve genital organlar gibi
ana organların anomalilerini içerebilir. Hastalığın bazı formları yenidoğan
döneminde ciddi şekilde kısıtlanmış torasik bir kafese ikincil solunum
yetmezliği nedeniyle öldürücüdür, diğerleri ise yaşamla uyumludur. Hastalık
spektrumu Ellis-van Creveld sendromunu, boğucu torasik distrofiyi (Jeune
sendromu), Mainzer-Saldino sendromunu, ve kısa kaburga-polidaktili sendrom
Belirtiler
Jeune sendrom öncelikle kemikleri etkileyen nadir bir durumdur. Bu
durumdan etkilenen insanlar tipik olarak iskelet anormallikleri ile
doğarlar:
Küçük, dar göğüs
Kısa kaburgalar
Kolların ve bacakların kısaltılmış kemikleri
Alışılmadık şekilli pelvis
Ekstra parmaklar ve / veya ayak parmakları
Jeune
sendromunun diğer özellikleri şunlardır; yüksek tansiyon, karaciğer hastalığı, pankreas
kistleri, diş anormallikleri ve göz hastalığı görme kaybına yol açabilecek retina
distrofisi denir.
Hastalar tipik olarak yenidoğan döneminde değişken derecelerde solunum
sıkıntısı ve tekrarlayan solunum yolu enfeksiyonları ile başvururlar. Bu
solunum problemleri ATD’nin en ciddi komplikasyonlarıdır ve bu hastalarda
mortalitenin ana nedenidir. Bazı raporlar ATD’li çocukların% 60-80’inin
bebeklik döneminde veya doğumdan sonraki ilk birkaç yıl içinde öldüğünü göstermektedir. Erken
çocukluk döneminde yaşayan hastalar için, solunum problemleri yaşla birlikte
iyileşme eğilimi gösterir, böylece bir hasta alt grubu ergenlik veya
yetişkinliğe yaşayabilir.
Çocuk büyüdükçe ATD’nin diğer komplikasyonları da olabilir: yüksek
tansiyon, böbrek kistleri, pankreas kistleri ve daha az yaygın karaciğer
hastalıkları, diş anormallikleri ve azalmış veya kötüleşen görme (retinal
distrofi).
Etkilenen bireyler, böbrek
yetmezliğine veya arızalara neden olabilecek kronik nefrit (böbrek rahatsızlığı)
geliştirebilir. Kalp anormallikleri ve hava yolunda daralma da
görülebilir.
Sebepleri
Birçok durumda, Jeune’nin sendrom nedeni bilinmeyen, ancak değişiklikler
(mutasyonlar) birkaç farklı genler( IFT80 , DYNC2H1 , WDR19 , IFT140 ve TTC21B ) bu durumdaki bazı
ailelerde tanımlanmıştır. Bu genlerin tümü,protein içinde bulunan hücresiliya adı verilen ve hücrelerin yüzeyinde
mikroskobik, parmak benzeri çıkıntılar olan yapılar . Kirpilerin
gelişimini ve bakımını bozan mutasyonların Jeune sendromu ile ilişkili belirti
ve semptomlara nasıl yol açtığı net değildir.
11 gendeki mutasyonların ATD’nin bugüne kadar neden olduğu
bulunmuştur. Genler şunlardır: CEP120, CSPP1, DYNC2H1, IFT80,
IFT140, IFT172, TTC21B, WDR19, WDR34, WDR35 ve WDR60 . Etkilenen
bireylerin yüzde 70’inin bu 11 genden birinde mutasyona sahip olduğu tahmin
edilmektedir. Bu genlerdeki mutasyonlar, kemik gelişimini etkileyen
anormal kirpikler proteinlerine yol açar.
ATD, otozomal resesif genetik bir hastalık olarak kalıtsaldır. Resesif
genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı
anormal geni miras aldığında ortaya çıkar. Bir kişi hastalık için bir
normal gen ve bir gen alırsa, kişi hastalık için bir taşıyıcı olacaktır, ancak
genellikle semptom göstermez. İki taşıyıcı ebeveynin hem değiştirilmiş
geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte%
25’tir. Her hamilelikte ebeveyn gibi taşıyıcı olan bir çocuk sahibi olma
riski% 50’dir. Bir çocuğun her iki ebeveynten normal gen alma şansı%
25’tir. Risk erkekler ve kadınlar için aynıdır.
Teşhis
Bazı durumlarda, Jeune tanısı sendromKarakteristik eğer doğumdan önce şüpheli
olabilir belirti ve bulgular mevcut olanultrason. Doğumdan sonra Jeune sendromuRöntgenBulgular. Bazı ailelerde, teşhis
ile doğrulanabilirgenetik test.
Genetik Test Kayıt (GTR) bu durum için genetik
testler konusunda bilgi sağlar. GTR için hedef kitle sağlık hizmeti
sağlayıcıları ve araştırmacılardır. Genetik test hakkında özel soruları
olan hastalar ve tüketiciler bir sağlık uzmanı veya bir genetik uzmanıyla
görüşmelidir.
Orphanet , bu durum için
tanısal testler sunan uluslararası laboratuvarları listeler.
Tedavi
Tedavi, solunum yolu enfeksiyonlarını
yönetmeye ve böbrek ve karaciğer fonksiyonlarını düzenli olarak izlemeye
dayanır. Şiddetli solunum yolu enfeksiyonu riski iki yaşından sonra
azalır.
Dikey genişletilebilir protez titanyum kaburga (VEPTR), pediyatrik
hastalarda torasik yetmezlik sendromunun (TIS) tedavisi için 2004 yılında FDA
tarafından onaylanmıştır. TIS, göğüs, omurga ve kaburgaların ciddi
deformitelerinin normal nefes almayı ve akciğer gelişimini engellediği
konjenital bir durumdur. VEPTR, omurganın düzeltilmesine ve kaburgaların
düzleştirilmesine yardımcı olan implante edilmiş, genişletilebilir bir
cihazdır, böylece akciğerler büyüyecek ve nefes almak için yeterli hava ile
doldurulabilir. Cihazın uzunluğu hasta büyüdükçe
ayarlanabilir. Spondilotorasik displazi tedavisi için, göğsün her iki
tarafında kaburgalar ayrılır ve göğsün her iki tarafına VEPTR’ler
yerleştirilir. Raynham Mass’ta DePuy Synthes Spine Co. tarafından
üretilmektedir.
Etolojisi
Sendromun moleküler temeli , her biri
bir intraflagellar taşıma proteinini kodlayan IFT80 (3q25.33), DYNC2H1 (11q22.3),
WDR19 (4p14) ve TTC21B (2q24.3) genlerinin dahil olduğunu gösteren kısmen
açıklanmıştır. Jeune sendromunun ciliopathies grubuna ait
olduğu. Diğer genlerdeki mutasyonlar da hastalığa karışabilir ve
tanımlanmaya devam edebilir.
Prognoz
Visseral ilişkili hastalıklara bağlı olarak
prognoz oldukça değişkendir ve ciddi solunum komplikasyonları riski 2 yaşından
sonra azalır.
Genellikle kalıtsal polipozis olmayan
kolorektal kanser (HNPCC) olarak adlandırılan Lynch sendromu, birçok
kanser türünün, özellikle kolon kanserlerinin riskini artıran kalıtsal bir hastalıktır. Lynch
sendromlu insanlar ayrıca mide, ince bağırsak, karaciğer, safra
kesesi kanalları, üst idrar yolu, beyin ve cilt kanseri riskinde artışa sahiptir.
Ek olarak, bu bozukluğu olan kadınların yumurtalık kanseri veya
rahim kanseri (endometrium) riski yüksektir. Bu bozukluğu olan
bireylerde, kolon polipleri (iyi huyluları) genel popülasyonda olduğundan daha
fazla görülür, ancak daha fazla sayıda olmaz.
Varyasyonlar MLH1 , Msh2 , Msh6 , PMS2 ve EPCAM-TACSTD1 delesyonlarını içeren fakat bunlarla sınırlı olmayan spesifik uyumsuzluk onarım (MMR) genlerinin mutasyonları Lynch sendromundan sorumludur.
MLH1 , MSH2 , Msh6 ,
ve Pms2 genlerinin herhangi birindeki mutasyonlar, DNA replikasyon
hatalarının düzgün onarımını önler. Anormal hücreler bölünmeye devam
ettikçe, biriken hatalar kontrolsüz hücre büyümesine ve muhtemelen kansere yol
açabilir. Ancak, Bu genlerdeki mutasyonlar bireyleri kansere yatkınlaştırsa da,
bu mutasyonları taşıyan herkes kanserli tümörler geliştirmez.
Lynch
sendromlu insanlar rutin kolonoskopi yaptırmalıdır.
Belirti ve Semptomlar
Aynı
hastalığı olan insanlar listelenen tüm semptomlara sahip olmayabilir. Karın
ağrısı, kabızlık, gastrointestinal kanamalar, yorgunluk başlıca belirtilerdir.
Lynch
sendromu nadir bir durum değil, daha çok teşhis konulan bir durumdur!
Bir kişi erken yaşlarda (50 yaş ve altı) kalın
bağırsak kanseri tanısı almış ve
Ailesinde farklı jenerasyonlarda 2’den fazla
kalın bağırsak vakaları görülmüş ise ya da
Ailesinde farklı jenerasyonlarda 2’den fazla
endometriyal, mide ve yumurtalık kanseri görülüyorsa,
Bu
bireylerde ve ailesinde Lynch sendrom için tarama genetik testler yapılmalıdır.
Genetik Görülme
Sıklığı
Amerika Birleşik Devletleri’nde her yıl
yaklaşık 140.000 yeni kolorektal kanser vakası teşhis edilir. Bu
kanserlerin yaklaşık yüzde 3 ila 5’ine
Lynch sendromu neden olur.
Kalıtım Paterni/ Deseni
Lynch sendromu kanser riski otozomal dominant bir paternde kalıtsaldır,
yani her hücrede değiştirilmiş genin kalıtsal bir kopyası kanser riskini
arttırmak için yeterlidir. İnsanların, hastalığın kendisini değil, artan
kanser riskini miras aldıklarını belirtmek önemlidir. Bu genlerde mutasyon
geçiren herkes kanser geliştirmez.
Lynch sendromlu bir ebeveynin çocukları% 50
mutasyon alma riskine sahiptir.
Teşhis Yöntemleri ve Tedavileri
Teşhis
Lynch sendromunu doğru bir şekilde teşhis etmek
için bilinen tek yöntem, bugün bilinen bir tedaviye en yakın olan genetik
testtir. Ailenin tıbbi öyküsü, ikisi doğrudan üçte biriyle ilişkili olan
ve her biri Lynch kanserlerini sürdüren üç aile üyesini gösteriyorsa, genetik
testler bir kişinin doktoru ile tartışılmalıdır.
Lynch sendromu teşhis edildikten sonra, yüksek
oranda hedeflenmiş bir tarama ve tıbbi yönetim programı gereklidir ve hayat kurtarıcı
olabilir. Rutin taraması sırasında, tümörler keşfedilebilir ve hayatı tehdit
etmeden önce daha kolay çıkarılır veya tedavi edilir.
Tedavi
Kolon kanseri tedavisi, kolonun etkilenen
kısmının cerrahi olarak çıkarılmasıdır (kolektomi).
Modern teknoloji ve özel araştırmacıların ve
tıp profesyonellerinin tutkulu ve özenli çabaları sayesinde. Henüz bir
tedavi bulunamamasına rağmen, genetik testlerle, ailelerde kanser riski
tanımlanabilir. Önleyici tedbirler (erken tanı, gözetleme ve tedavi)
uygulanarak, daha yüksek yaşam kalitesi ve uzun ömür elde edilebilir ve
bireyler ve aileler kanserden korunabilir. Genetik testler ve yıllık taramalar
bizim için bir tedaviye en yakın şeydir.
Genetik yatkınlıkla teşhisi yapılan Lynch
sendromlu kişiler, kolon kanserine yatkınlığın riskini çevresel faktörlerle
arttırmamak için, öncelikle sigaradan uzak durmalı. Sağlıklı beslenme
alışkanlıklarını hayatına katarak, stresten uzak kalmaya özen göstermelidir.
Juvenil
hemokromatoz, vücudun çeşitli organlarında demir birikimi ile karakterize nadir
görülen bir genetik bozukluktur. Klasik kalıtsal hemokromatozdan ayrı, farklı
bir hastalıktır. Juvenil hemokromatoz, farklı genlere mutasyonlardan
kaynaklanır ve genellikle daha erken bir başlangıç yaşı ve daha şiddetli
demir birikimine sahiptir.
Hemokromatoz
tip 2 belirtileri tipik olarak çocukluk döneminde başlar. Hemokromatoz tip
2’nin semptomları genellikle 30 yaşından önce belirginleşir.
Genetik Faktörler/ Etken Faktörler
İki genin (HJV
ve HAMP) mutasyonlarının juvenil hemokromatoza neden olduğu bilinmektedir. HJV
genindeki mutasyonlar, bilinen çocuk hemokromatoz vakalarının yüzde 90’ından
fazlasını oluşturmaktadır. HJV ve HAMP genlerinin mutasyonları, otozomal resesif
özelliklerdedir. Genetik hastalıklar, babadan ve anneden alınan kromozomlarda
bulunan belirli bir özellik için genlerin kombinasyonu ile belirlenir.
Tip 2A, en sık
görülen form, kromozom 1 üzerindeki hemojuvelin ( HJV ) genindeki
mutasyonlardan kaynaklanır ve tip 2B, kromozom 19 üzerindeki hepsidin ( HAMP )
genindeki mutasyonlardan kaynaklanır. Bu mutasyonlar, hepsidin eksikliğine neden olur, böylece
dalaktan büyük ölçüde duodenum demir emilimi ve demir salınımının artmasına
neden olur.
Belirti ve Semptomlar
Juvenil
hemokromatozun semptomları ve şiddeti kişiden kişiye değişebilir. Başlıca
belirtileri kalp, karaciğer ve eklem hastalığıdır. Bazı durumlarda, spesifik
olmayan, belirsiz semptomlar daha ciddi komplikasyonların gelişmesinden önce
gelebilir. Bu semptomlar yorgunluk, eklem ağrısı (artralji) ve iştahsızlığı
içerebilir. Tedavi edilmezse, çocuk hemokromatozu , hayatı tehdit eden
komplikasyonlara neden olabilir.
Juvenil
hemokromatoz belirtileri genellikle 30 yaşından önce belli bir noktada ortaya
çıkar. Bununla birlikte, nadir durumlarda, bazı kişiler 30’larına kadar semptom
geliştirmeyebilir.
Juvenil
hemokromatoz ile ilişkili yaygın bir semptom olan hipogonadotropik
hipogonadizm, kadınlarda yumurtalıkların veya erkeklerde veya testislerin yok
veya azalmış fonksiyonu ile karakterizedir. Hipogonadotropik hipogonadizm, daha
önce adet görmeye başlayan kızlarda altı ay boyunca adet döngüsünün azalmasına
veya yokluğuna, ergenliğe ulaşmada gecikmelere, genitalde ve koltukaltlarında
kıllanmada azalma, erkeklerde iktidarsızlığa neden olabilir. Cinsel isteksizlik
ve kısırlık da bu durumla ilişkili olabilir. Uzun süreli hipogonadizm, düşük
kemik yoğunluğuna (osteopeni) ve kırığa eğilimli kemiklere (osteoporoz) neden
olabilir.
Juvenil
hemokromatozlu birçok bireyde kalp kası hastalığı (kardiyomiyopati)
gelişebilir. Kalp anormallikleri aniden başlayabilir ve düzensiz kalp atışları
(aritmiler) ve kalp yetmezliği gibi ciddi, hayatı tehdit eden komplikasyonlara
neden olabilir. Bazı durumlarda, kalp hastalığı, çocuk hemokromatozunun ilk
belirtisi olabilir.
Juvenil
hemokromatoz ile potansiyel olarak ilişkili ek semptomlar arasında, cilt
lekelerinin (artmış cilt pigmentasyonu), eklem hastalığının (artropati) ve
karaciğer hastalıkları(hepatomegali ve siroz) ortaya çıkması yer alır.
Juvenil
hemokromatozu olan bireylerde demir birikimi pankreasta da ortaya çıkabilir.
Pankreas, midenin arkasında, bağırsaklara giden ve sindirime yardımcı olan
enzimleri salgılayan küçük bir organdır. Pankreas ayrıca şekeri parçalamaya
yardımcı olan insülin gibi diğer hormonları da salgılar. Pankreastaki hasar
diyabetes mellitusa yol açabilir. En belirgin semptomlar alışılmadık derecede
aşırı susuzluk ve idrara çıkmadır.
Genetik Görülme Sıklığı
Hemokromatoz tip
2, geniş bir coğrafi dağılıma sahip nadir bir hastalıktır. Her iki cinsiyet de
eşit derecede etkilenir. Bozukluk nadirdir, ancak genel popülasyondaki gerçek
insidansı bilinmemektedir. Bozukluk
genellikle 10-30 yaşları arasında görülür. HJV genindeki mutasyonlar, genç
hemokromatoz vakalarının çoğunu oluşturur.
Kalıtım Paterni/Deseni
Hemokromatoz tip
2, otozomal resesif olarak kalıtılmaltadır.
Teşhis Yöntemleri
Juvenil hemokromatozlu bireylerde erken tanı
ve acil tedavi şarttır ve aşırı demir depolamasından kaynaklanan kalıcı organ
hasarını ve potansiyel olarak hayatı tehdit eden komplikasyonları önlemeye
yardımcı olabilir. Bozukluk belirli fiziksel bulguların (hepatomegali, diabetes
mellitus, anormal cilt pigmentasyonu, kalp hastalığı, hipogonadizm ve / veya
artrit dahil) saptanması, kapsamlı bir hasta öyküsü; tam bir aile öyküsü ve
özel testler gibi kapsamlı bir klinik değerlendirmeye dayanarak teşhis edilebilir.
Juvenil
hemokromatozdan şüphelenildiğinde, kandaki anormal derecede artmış demir
seviyelerini tespit etmek için kan testleri yapılır; vücudun demir depolarının
bir göstergesi olarak kullanılan bir demir bileşiğinin yüksek kan seviyeleri
(serum ferritin seviyeleri); ve artan transferrin doygunluğuna bakılır.
(Transferrin, demirin bağırsaktan kan dolaşımına taşınmasında rol oynayan bir
proteindir.)
Ek olarak,
manyetik rezonans görüntüleme (MRI) gibi özel görüntüleme testleri, aşırı demir
birikimi nedeniyle karaciğerin artan yoğunluğunu ortaya çıkarabilir. MRI,
belirli organ ve dokuların ayrıntılı kesit görüntülerini sağlamak için manyetik
alan ve radyo dalgaları kullanır. Juvenil hemokromatozis tanısında yardımcı
olmak için karaciğer biyopsisi de kullanılabilir. Karaciğer biyopsisi
sırasında, artan demir depolamasını ve siroz varlığını tespit etmek için
karaciğer doku örnekleri alınır ve mikroskopik olarak incelenir.
Juvenil
hemokromatoz tanısı, bozukluğa neden olan HJV veya HAMP genlerinin karakteristik
mutasyonlarını ortaya çıkarabilen moleküler genetik test ile doğrulanabilir.
Moleküler genetik test klinik olarak yapılabilir.
Tedaviler
Juvenil
hemokromatozis tedavisi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Tedavi genellikle klasik hemokromatozis için mevcut tedavi
seçeneklerine benzer. Temel olarak, doktorlar flebotomi adı verilen bir
prosedürle vücuttaki fazla demiri çıkaracaklardır.
Vücuttaki demirin
çoğu kırmızı kan hücrelerinde bulunur. Bu nedenle, terapi, vücudun içinden
fazla demiri azaltmak için kanın bir damar yoluyla düzenli olarak çıkarılmasını içerir.
Flebotomi haftada bir veya iki kez gerekebilir. Kabul edilebilir demir
seviyelerine ulaşıldığında, haftalık flebotomi tedavisi durdurulur ve idame
tedavisi başlatılır. İdame tedavisi ile bireyler kanı (demir seviyelerini
azaltmak için) her hafta verir. Gereken spesifik miktar değişir, ancak
genellikle yılda dört ila altı flebotomi yeterlidir.
Demir
şelatörleri genellikle aşırı demir yükünün diğer bozukluklarını tedavi etmek
için kullanılsa da, çocuk hemokromatozu olan kişiler için önerilmez. Demir
şelatörleri, vücuttaki fazla demire bağlanan ve suda çözünmesine ve böbrekler
yoluyla vücuttan atılmasına izin veren ilaçlardır. Deferoksamin, anemi veya
ciddi kalp hastalığı olan çocuk hemokromatozu olan bireylerde ek tedavi olarak
kullanılan bir demir şelatörüdür.
Çocuk
hemokromatozunun erken, hızlı tespiti ve tedavisi önemlidir, çünkü organ
hasarını ve bazı ikincil komplikasyonların gelişmesini önleyebilir. Çocuklukta
erken teşhis konan ve flebotomi programlarına yerleştirilen ve bunlara bağlı
olan bireylerde ikincil komplikasyonların gelişimi önemli ölçüde azalmıştır. Gelişen
ikincil komplikasyonlar standart, geleneksel yöntemlerle tedavi edilir.
Hipogonadotropik
hipogonadizm, hormon replasman tedavisi ile tedavi edilebilir. Eklem ağrısı
steroidal olmayan anti-enflamatuarlarla tedavi edilebilir. Kalp hastalığı,
anjiyotensin dönüştürücü enzim (ACE) inhibitörleri, vücuttan tuz ve suyun
çıkarılmasına yardımcı olan ilaçlar (diüretikler) ve kalp yetmezliği ve
aritmilerin tedavisinde yaygın olarak kullanılan glikozitler adı verilen
ilaçlarla tedavi edilir. Bazı durumlarda, şiddetli, geri döndürülemez kalp
hasarında kalp nakli gerektirir.
Fibroz gibi
karaciğer hastalığının erken belirtileri flebotomi ile tedavi edilebilir.
Klasik hemokromatozlu bazı bireylerde, bu semptomlar geri dönüşümlüdür. Juvenil
hemokromatozu olan bireylerde bu semptomların geri dönüşümlü olup olmadığı
bilinmemektedir. Karaciğer sirozu geri dönüşümlü değildir ve propranolol ve
nadolol ile tedavi gerektirir. Bazı durumlarda, karaciğer nakli gerekli
olabilir.
Genetik
danışmanlık etkilenen bireyler ve aileleri için yararlı olabilir. Diğer tedavi
semptomatik ve destekleyicidir.


{kind=link}