Genel
Bilgi, Genetik Değişiklikler/Etken Faktörler
2-hidroksiglutarik asidüri, beyne ilerleyen
hasara neden olan bir durumdur. Bu bozukluğun ana tiplerine
D-2-hidroksiglutarik asidüri (D-2-HGA), L-2-hidroksiglutarik asidüri (L-2-HGA)
ve kombine D, L-2-hidroksiglutarik asidüri (D, L) denir. -2-HGA). D-2-HGA’nın
ana özellikleri farklı tipler arasında farklılık gösterir, ancak genel olarak
gecikmiş gelişimi içerebilir; nöbetler; zayıf kas tonusu (hipotoni); ve kas
hareketi, konuşma, görme, düşünme, duygu ve hafıza gibi birçok önemli işlevi
kontrol eden beynin en büyük bölümündeki anormallikler. Farklı tiplere ve alt
tiplere farklı gen mutasyonları neden olur ve otozomal dominant bir desende
kalıtsal olarak tip II D-2-HGA olarak bilinen bir D-2HGA alt tipi hariç,
otozomal resesif bir paternde kalıtsaldır. henüz tedavi. Tedavi semptomlara
bağlıdır. Yönetim esas olarak nöbetlerin mevcut olduklarında kontrol edilmesini
içerir.
D2HGDH ve L2HGDH genleri,
hücreler içindeki enerji üretim merkezleri olan mitokondride bulunan enzimlerin
yapılması için talimatlar sağlar. Enzimler, hücresel aktiviteler için enerji
üreten bir dizi reaksiyonun bir parçası olarak, sırasıyla D-2-hidroksiglutarat
ve L-2-hidroksiglutarat adı verilen bileşikleri parçalamaktadır. Bu genlerin
herhangi birindeki mutasyonlar, hücrelerde D-2-hidroksiglutarat veya
L-2-hidroksiglutarat birikmesine yol açan fonksiyonel enzim eksikliğine yol
açar. Yüksek seviyelerde, bu bileşikler hücrelere zarar verebilir ve hücre
ölümüne yol açabilir. Beyin hücreleri, bu bileşiklerin toksik etkilerine karşı
en savunmasız gibi görünmektedir, bu da D-2-HGA tip I ve L-2-HGA’nın belirti ve
semptomlarının neden beyni kapladığını açıklayabilir.
SLC25A1 geni, sitrat gibi
belirli molekülleri mitokondriya içine ve dışına taşıyan bir proteinin üretimi
için talimatlar sağlar. SLC25A1 genindeki mutasyonlar protein fonksiyonunu
azaltır, bu da D-2-hidroksiglutarat ve L-2-hidroksiglutarat birikmesine ve
beyin hücrelerine zarar verir. Araştırmacılar, özellikle sitrat olmak üzere
diğer moleküllerin dengesizliğinin de kombine tipte belirti ve semptomlara
katkıda bulunduğundan şüpheleniyor.
IDH2 geninde mutasyonlar
olduğunda, normal aktivitesini gerçekleştirmesini engelleyen değiştirilmiş bir
enzim oluşur, (fonksiyonu: izositratın 2-ketoglutatata dönüştürülmesi). Bunun
yerine, değiştirilmiş enzim yeni bir anormal fonksiyon alır:
D-2-hidroksiglutarat adı verilen bir bileşiğin üretilmesi. Fazla
D-2-hidroksiglutarat toksiktir ve beyin hücrelerine zarar vererek D-2-HGA tip
II’nin belirti ve semptomlarına yol açar.
D-2- hidroksiglutarik asidüri (D-2-HGA),
gelişimsel gecikme, nöbetler, zayıf kas tonusu (hipotoni) ve beyindeki
anormalliklerle ve kas hareketi, konuşma, görme, düşünme, duygular ve bellek
ile karakterize edilir.
Tip I ve tip II olmak üzere iki D-2-HGA alt
tipi vardır. İki alt tip, genetik nedenleri ve kalıtım şekli ile ayırt edilir,
ancak işaret ve semptomlarda da bazı farklılıklar vardır. Tip II daha erken başlama eğilimindedir ve tip
I’den daha şiddetlidir ve zayıflamış ve genişlemiş bir kalp (kardiyomiyopati)
ile ilişkili olabilir.
L-2-hidroksiglutarik asidüri (L-2-HGA)
özellikle beynin beyincik denilen, hareketlerin koordinasyonunda rol oynayan
bir bölgesini etkiler. Sonuç olarak, etkilenen insanlar denge ve kas
koordinasyonu (ataksi) ile ilgili problemlere sahiptir. L-2-HGA’nın diğer
özellikleri arasında gelişimsel gecikme, nöbetler, konuşma güçlükleri ve aşırı
büyük bir kafa (makrosefali) sayılabilir. Belirti ve semptomlar ve şiddetleri
değişkendir ancak genellikle bebeklik veya erken çocukluk döneminde başlar ve
zamanla kötüleşir ve genç yetişkinlerde ciddi sakatlıklara neden olur. Kombine
tip D, L-2-hidroksiglutarik asidüri (D, L-2-HGA) erken çocukluk döneminde
başlayan ciddi beyin bozukluklarına neden olur. Etkilenen çocukların şiddetli
nöbetleri, zayıf kas tonusu (hipotoni), beslenme sorunları ve çok ciddi solunum
problemleri vardır.
2-Hidroksiglutarik asidüri nadir görülen bir
hastalıktır. D-2-hidroksiglutarik asidüri (D-2-HGA) ve L-2-hidroksiglutarik
asidüri (L-2-HGA) dünya çapında yaklaşık 150 kişide rapor edilmiştir. Kombine
tip D, L-2-HGA daha nadir görülür ve yaklaşık bir düzine bildirilen vaka
vardır.
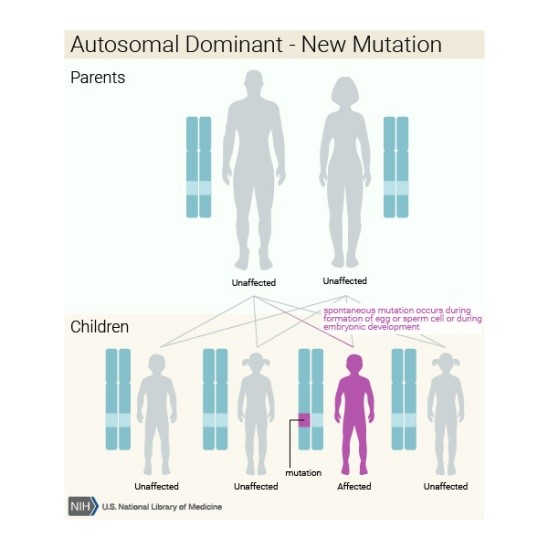
D-2-HGA tip I, L-2-HGA ve
kombine D, L-2-HGA tiplerinin otozomal resesif kalıtım şekli vardır.Tip D-2-HGA
alt tipi II, otozomal dominant bir hastalık olarak kabul edilir. Bu tip
genellikle IDH2 geninde yeni bir değişikliğin (de novo mutasyon) bir sonucudur
ve aile öyküsü olmayan kişilerde görülür.
Semptomları gösteren hastalar için birkaç tip klinik
test yapılabilir; Biyokimyasal Genetik Testleri (Analit), Moleküler Genetik
Testleri (Hedeflenen varyant analizi, Silme / çoğaltma analizi, Seçili
eksonların sekans analizi, Tüm kodlama bölgesinin sekans analizi)
Halen yerleşik bir tedavi yoktur ancak
nöbetler gibi bazı semptomlar ilaçla tedavi edilebilir. Bununla birlikte, D,
L-2HGA tipi (birleşik tip) için bazı araştırmalarda malat veya sitrat
kullanılmıştır. Malat tedavisi sırasında üriner malat konsantrasyonu artmış,
ancak bunun ötesinde hiçbir etki gözlenmemiştir. sitrat ile tedavi ise bazı iyi
sonuçlara yol açtı ancak bu tedavinin etkinliğini doğrulamak için daha fazla
çalışmaya ihtiyaç duyuluyor. Tıbbi literatürdeki olgu sunumları bazı
tedavilerin bazı durumlarda etkili olabileceğini göstermiştir.
Farklı 2-hidroksiglutarik asitüri tipleri,
çeşitli genlerdeki mutasyonlardan kaynaklanır. D-2-HGA tip I, D2HGDH genindeki
mutasyonlardan kaynaklanır; tip II, IDH2 genindeki mutasyonlardan kaynaklanır.
L-2-HGA, L2HGDH genindeki mutasyonlardan kaynaklanır. Kombine tip D, L-2-HGA,
SLC25A1 genindeki mutasyonlardan kaynaklanır.
Hemokromatoz
tip 4 olarak da bilinen ferroportin hastalığı, vücutta anormal demir birikimi ile karakterize
nadir görülen bir genetik bozukluktur. Ferroportin hastalığına SLC40A1 geninin
mutasyonları neden olur. Hemokromatoz tip 4 iki
alt tipe ayrılabilir:
Hemokromatoz tip 4A
Hemokromatoz tip 4B
Hemokromatoz tip 4A olan kişilerde hastalığın
herhangi bir belirtisi olmayabilir. Bireyler yaşlandıkça
karaciğer hastalığı geliştirebilirler. Hemokromatoz tip 4B, yorgunluk,
halsizlik ve eklem ağrısı ile belirti gösterebilir. Diğer semptomlar arasında karın ağrısı, cinsel dürtü kaybı, karaciğer
hastalığı, diyabet, kalp problemleri, nefes almada güçlük ve ciltte renk değişikliği
sayılabilir. Hemokromatoz tip 4B semptomları çocukluktan yetişkinliğe kadar her zamanda başlayabilir.
Genetik Değişiklikler/Etken
Faktörler
Ferroportin
hastalığına SLC40A1 geninin mutasyonları neden olur. SLC40A1 geni, demirin
uygun şekilde hücrelerden dışarı aktarılması için kritik olan özelleşmiş protein ferroportini oluşturmak
için bilgi içerir. Ferroportin ayrıca demirin uygun
şekilde parçalanmasında (metabolizma) rol oynar. Demir, vücudun tüm hücrelerinde
bulunan ve vücudun düzgün çalışması ve büyümesi için gerekli olan kritik bir
mineraldir. Demir, kırmızı et, kümes hayvanları, yumurta ve sebzeler de dahil
olmak üzere birçok yiyecek türünde bulunur. Demir seviyeleri vücut içinde
belirli bir aralıkta kalmalıdır, aksi takdirde anemiye (düşük demir seviyelerinden
dolayı) veya etkilenen organlara (yüksek demir seviyelerinden dolayı) zarar
verebilir.
SLC40A1 geninin mutasyonları, düşük
seviyelerde fonksiyonel ferroportin ile sonuçlanır. Fonksiyonel ferroportin
eksikliği sonuçta vücudun hücrelerinde ve dokularında anormal demir birikmesine
neden olur. SLC40A1 geninin farklı mutasyonları , ferroportin proteinini farklı
şekillerde etkiler, bu nedenle demirin hücre dışına aktarılmasını ve metabolizmasını değiştirir. Araştırmacılar, SLC40A1 mutasyonlarının
ferroportini etkilemesinin farklı yollarının , bozukluğun iki farklı formunu
oluşturduğu düşünülmektedir.
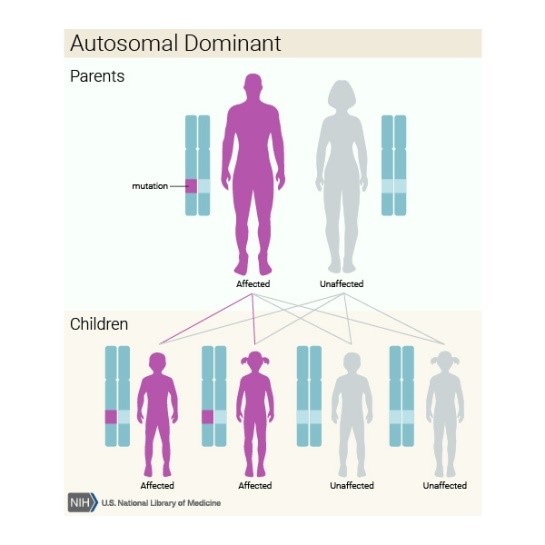
Ferroportin hastalığı, otozomal dominant
genetik bir durum olarak kalıtsaldır. Baskın genetik bozukluklar, hastalığın
ortaya çıkması için anormal bir genin sadece tek bir kopyası gerektiğinde
ortaya çıkar. Anormal gen, her iki ebeveynden miras alınabilir veya etkilenen
bireyde yeni bir mutasyonun (gen değişimi) sonucu olabilir. Anormal geni
etkilenen ebeveynden yavrulara geçirme riski, her hamilelik için yüzde 50’dir.
Risk erkekler ve kadınlar için aynıdır.
Belirti ve
Semptomlar
Genellikle ferroportin hastalığı iki ana forma
ayrılır.
Ferroportin hastalığı olan bazı kişilerde
hafif bir bozukluk şekli gelişir. Bu bireylerin kan plazmasında yüksek
seviyelerde ferritin (hiperferritinemi) ve düşük düzeyde doymuş transferrin
(kanda demir taşıyan protein) bulunur. Etkilenen bireyler yaşlandıkça, hafif
karaciğer hasarı (hepatik fibroz) ortaya çıkabilir.
Diğer bireyler, hemokromatozun (hemokromatoz
tip 1) daha yaygın klasik formuna benzeyen bir ferroportin hastalığı formu
geliştirir. Transferrin doygunluğu bu formda önemli ölçüde yükselir. Bu formla
ilişkili semptomlar arasında eklem ağrısı, kalbin ritmindeki anormallikler veya
kalp atışı paterni (aritmiler) ve diyabet bulunur. Karaciğer hasarı bu
ferroportin hastalığı formunda daha yaygındır ve karaciğerin skarlaşmasına
(siroza) neden olabilir.
Klinik
Belirti ve Bulgular
Çok sık
Artralji (eklem ağrısı)
Genelleştirilmiş
hiperpigmentasyon
Artan serum ferritin
Eklem çıkığı
Eklem şişmesi
Sık
Karın ağrısı
Hepatik steatoz
Nadiren
Siroz
Konjenital hepatik fibroz
Genetik Görülme Sıklığı
Ferroportin
hastalığı erkekleri ve kadınları eşit sayıda ve tüm ırklardan ve etnik kökenlerden bireyleri
etkiler. Hemokromatoz tip 4 en çok güney Avrupa kökenli
insanlarda yaygındır. Ferroportin
hastalığının kesin insidansı bilinmemekle birlikte <1/1.000.000 olduğu düşünülmektedir.
Kalıtım
Paterni/Deseni
Hastalık otozomal dominant bir şekilde kalıtsaldır.
Hastalığın
Diğer İsimleri
HFE4, hemokromatoz otozomal
dominant, ferroportin defekti nedeniyle hemokromatoz, otozomal dominant kalıtsal hemokromatoz, ferroportin hastalığı
Hastalıkla İlişkili
Genler
SLC40A1
geni
Teşhis Yöntemleri
Ferroportin
hastalığının teşhisi, karakteristik semptomların tanımlanması, ayrıntılı bir
hasta öyküsü, kapsamlı
bir klinik değerlendirme ve çeşitli özel testlere dayanarak yapılır. Kan
testleri, kandaki yüksek seviyelerde ferritin ve hastalığın daha hafif
formunda, transferrin düşük veya normal doygunluğu da dahil olmak
üzere ferroportin hastalığı ile ilişkili belirli bulguları, demirin vücutta doğru
taşınmasında rol oynayan başka bir proteini ortaya çıkarabilir. SLC40A1 geninin
mutasyonları için moleküler genetik testler mevcuttur ve tanıyı doğrulamak için
gereklidir.
Tedaviler
Hemokromatoz tip 4 tedavisi, etkilenen kişinin
hemokromatoz tip 4A veya hemokromatoz tip 4B olup olmamasına bağlıdır.
Hemokromatoz tip 4B olan kişiler, diğer hemokromatozis türlerine benzer şekilde
tedavi edilebilir. Tedavi seçenekleri arasında flebotomi yöntemiyle(kanın bir damar yoluyla çıkarılması) demir seviyelerinin düşürülmesi, demir şelasyon tedavisi(vücuttaki fazla demire bağlanan ve suda çözünmesine izin veren ve
böbrekler yoluyla vücuttan atılan ilaçlardır) diyet değişiklikleri ve
hastalığın komplikasyonlarının tedavisi yer alabilir.
Hemokromatoz tip 4A olan kişilerin tedavisi
tipik olarak flebotomi içermez. Çünkü hemokromatoz tip 4A olan insanlar,
flebotomi tedavileri alırlarsa anemi(düşük seviyede kırmızı kan hücresi) gelişebilir. Bunun yerine, tip 4A
hemokromatozlu insanlar, hastalığın semptomlarına sahip olup olmadıklarını ve
hastalığın en iyi nasıl tedavi edilebileceğini belirlemek için kan testleri ile
yakından izlenir.
Son olarak
hemokromatozlu insanlar
için diyet önerileri alkol ve kırmızı etten kaçınmayı içerebilir.
Hemokromatozlu kişilerin demir veya C vitamini takviyesi almaları önerilmez.
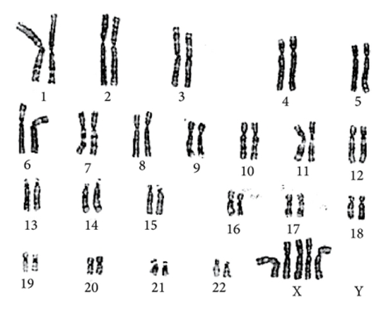
Penta X
sendromu kadınları etkileyen bir nadir cinsiyet kromozom anomalisidir. Dişilerde normalde iki X kromozomu bulunur. Pentasomy X sendromunda ise, kadınlarda üç
ekstra X kromozomu bulunur. (46, XX yerine 49, XXXXX).
Sendrom genel olarak; şiddetli zihinsel gerilik, gelişimsel
gerilik, kısa boy, kafatası ve yüz bölgesi kusurları ve diğer fiziksel
anormalliklerle karakterizedir. El ve ayaklar genelde küçüktür. Karakteristik
yüz kusurları; yukarı doğru uzanangöz kapağı kıvrımlarını, düz burun
kemiklerini, yanlış şekillenmiş kulakları, kısa bir boynu içerebilir.
Pentasominin X’in mayoz bölünme sırasında art arda maternal
disfonksiyonun bir sonucu olarak ortaya çıktığı düşünülmektedir.
Penta
X Sendromu, üç ekstra X kromozomunun varlığı ile karakterize bir kromozomal
bozukluktur. İnsan kromozomu çiftleri 1’den 22’ye kadar numaralandırılır,
normalde erkekler için bir X ve bir Y kromozomundan ve kadınlar için iki X
kromozomundan oluşan 23. çift (46 kromozom) bulunur.
Penta
X Sendromlu kadınlarda beşi X kromozomu olan 49 kromozom vardır (49, XXXXX
karyotipi). Fazladan üç X kromozomunun varlığı, ebeveynlerden birinde üreme
hücrelerinin bölünmesi sırasındaki hatalardan kaynaklanır (mayoz bölünme
sırasında ayrılmama).
Araştırmalar,
ekstra X kromozomlarının tipik olarak anneden alındığını göstermektedir. Buna
ek olarak, araştırmacılar bu tür hata riskinin ileri ebeveynlik yaşıyla
artabileceğini belirtmektedir.
Belirti
ve Semptomlar
Penta
X Sendromu karakteristik olarak doğum öncesi büyüme gecikmeleri ile
ilişkilidir. Ayrıca doğum sonrası büyüme ve kilo alma geriliği (gelişememe) ve
kısa boy gözlenir. Ek olarak Penta X sendromlu bebek ve çocuklarda genellikle
psikomotor becerilerin kazaılmasında gecikme gözlenir.
Bozukluk
ayrıca kafatası ve yüz (kraniyofasiyal) bölgesinin belirgin malformasyonları
ile karakterizedir. Bunlar genellikle alışılmadık derecede küçük bir kafa
(mikrosefali); yuvarlak bir yüz; yukarı eğimli göz kapağı kıvrımları (palpebral
fissürler); düz burun kemiği; yanlış biçimlendirilmiş kulaklar; ve düşük saç
çizgisine sahip kısa bir boyundur.
Bazı
durumlarda, Penta X Sendromu doğumda kalbin belirli yapısal kalp kusurları ile
ilişkili olabilir. Penta X Sendromlu bazı kadınlarda ayrıca böbrek
anormallikleri görülür.
Görülme
Sıklığı
Prevalans bilinmemektedir, ancak literatürde şimdiye kadar 40’tan
az vaka tanımlanmıştır.
Teşhis
Yöntemleri ve Tedaviler
Penta
X Sendromunun teşhisi, karakteristik fiziksel bulguların tespiti; ve vücut
hücrelerinde üç ekstra X kromozomunun varlığını doğrulayan kromozomal analiz
ile gerçekleşir. Bazı durumlarda, anormallik doğumdan önce (doğum öncesi
olarak) amniyosentez veya koryonik villus örneklemesi (CVS) gibi belirli
prosedürleri takiben kromozom analizine dayanarak tespit edilebilir.
Penta
X Sendromunun tedavisi, her bir kişide belirgin olan spesifik semptomlara
yöneliktir. Kişide görülen kusurların cerrahi olarak düzeltilmesini içerebilir.
Ek olarak, konjenital kalp kusurları olanlar için, bazı ilaçlarla tedavi,
cerrahi müdahale ve / veya diğer önlemler gerekebilir. Gerçekleştirilen
spesifik cerrahi prosedürler, anatomik anormalliklerin ciddiyetine ve konumuna,
ilişkili semptomlarına ve diğer faktörlere bağlı olacaktır.
Etkilenen
çocukların potansiyellerine ulaşmasını sağlamak için erken müdahale önemli
olabilir. Yararlı olabilecek özel hizmetler arasında özel eğitim, fizik tedavi
ve diğer tıbbi, sosyal ve mesleki hizmetler yer alır.
Ritscher-Schinzel sendromu olarak da bilinen 3C sendromu, kraniyofasiyal anormallikler, konjenital kalp kusurları ve serebellar beyin malformasyonları görülen gelişimsel bir malformasyon sendromudur.
Klinik Tanım
3C sendromu, azalan sıklıkla, alçak konumlu kulaklar, hipertelorizm,
aşağı eğik palpebral fissürler, basık burun köprüsü, belirgin oksiput, belirgin
alın, mikroginati, oküler kolobom, yarık damak gibi belirgin kraniyofasiyal
özellikler ile taşıyan konjenital bir hastalıktır. Ek kraniyofasiyal özellikler,
nevus flammeus’u (alın bölgesini) kapsar.
arka saç çizgisini, seyrek kafa derisi saçları, kaşları ve kirpikleri,
çıkıntılı dille açık ağız ve kısa boyunu içerir. Olguların% 80’inde serebellar
anomaliler vardır ve bunlar arasında öncelikle serebellar vermis hipoplazisi ve
sisterna magmanın genişlemesi yer alır. Etkilenen bireylerde hantal motor,
zihinsel engel ve konuşma gecikmesi vardır. Kardiyovasküler anomaliler arasında
atriyal ve ventriküler septal kusurlar, patent arteriyel kanal, Fallot
tetralojisi, çift çıkışlı sağ ventrikül, hipoplastik sol kalp sendromu , aortik
veya pulmoner stenoz ve diğer kapak anomalileri bulunur. Birçok hastada doğum
sonrası kısa boyluluk ve 2 hastada da büyüme hormonu eksikliği rapor
edilmiştir. Hastaların >%10’unda görülen ek anomaliler arasında beslenme
güçlükleri, tek göbek arteri, tek enine kırışıklı küçük eller, kamptodaktili,
ekinovarus bozukluğu, hidronefroz, yüzeysel skrotum, inmemiş testis, kriptorşidizm,
mikropenis, hipospadias, tırnak hipoplazisi, işitme kaybı, bağırsakta
malrotasyon. İskelet kusurları kaburga ve vertebral anomaliler ile ortaya
çıkabilir. Nadiren gözlenen özellikler arasında oküler (konjenital glakom, göz
kapağı pitozu ile optik sinir atrofisi, heteokromatik iris, posterior embriyotokson),
böbrek (multisistik displastik böbrek, at nalı böbrekler, tek taraflı böbrek
agenezi) ve anal (anal atrezi, anterior yerleşimli anüs malformasyonları)
anomalileri vardır. Renal hipoplazi, meme başı hipoplazisi, penil hipoplazi,
tek taraflı adrenal aplazi,immün yetmezlik rapor edilmiştir.
Epidemiyolojisi
Bugüne kadar <50 vaka tanımlanmıştır. Sendrom panetnik görünüyor.
Kalıtım Modeli
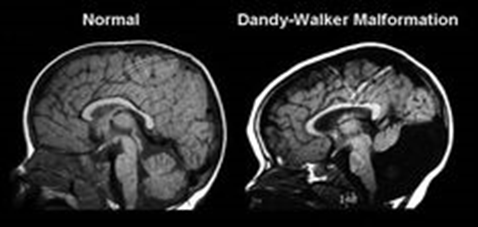
Dandy-Walker malformasyonu vakalarının çoğu sporadiktir, bu da
ailelerinde kusurlu olma durumunun olmadığı kişilerde ortaya çıktığı anlamına
gelir. Vakaların küçük bir yüzdesi ailelerde de görülüyor ancak, Dandy-Walker
malformasyonunun açık bir miras modeli yoktur. Birden fazla genetik ve çevresel
faktör muhtemelen bu bozukluğun gelişme riskini belirlemede rol oynamaktadır.
Dandy-Walker malformasyonuna sahip kişilerin birinci dereceden akrabalarında
(kardeşler ve çocuklar gibi) genel popülasyondaki insanlara kıyasla daha yüksek
oranda görülür.
Yönetim ve Tedavi
Yönetim esas olarak eğitim programları, fiziksel, mesleki ve konuşma
terapisini içeren semptomatik ve multidisipliner yaklaşımlardır. Hipotoni ve gelişmeye
yönelikmotor gecikmesini azaltmayı güçlendirmek için yapılan kardiyak
malformasyonlar özel bakım gerektirir, genellikle ameliyattır.
Prognoz
Prognoz kardiyovasküler malformasyon ile belirlenir. Motor gecikmesi
yaygındır ve serebellar anomalilere sekonder hipotoni ile ilişkilidir.
Hastalıkla İlgili Genler
3C/Ritscher-Schinzel sendromu doğuştan kraniyo-serebello-kardiyak
displazi’ye sebep olan, 11.kromozomunda
görülen mutasyona sebep olan CCDC22 ve WASHC5 genleriyle alakalıdır.
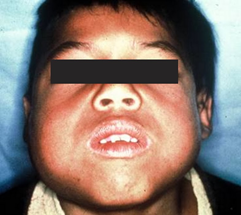
Cherubism (MIM 118400), üst çene (maxilla)
ve/veya alt çene (mandibula) kemiklerinin aşırı bozunması
ile tanımlanan karakteristik bir yüz şişmesine
neden olan, fibröz doku kitlelerinin gelişmesiyle
takip edilen kalıtsal otozomal dominant bir sendromdur (1).
İlk defa 1933 yılında Kanadalı radyolog
William Jones tarafından yanakların şişmesiyle
beraber, ırsi multiloküler (Çok sayıda küçük
boşluklar
gösteren) kistik bir çene hastalığı olarak tanımladı (2). Özellikle
üstçenedeki genişleme, yüz derisini ve alt göz kapağını aşağı doğru
çeker; hastanın yüzünde gökyüzüne bakıyormuş gibi
bir izlenimi ortaya çıkar. (Hastanın yüz ifadesi, orta çağ
tablolarındaki “meleklere bakan çocuklara” benzetilmiş ve
cherubism nitelemesi bu algılamadan doğmuştur.)
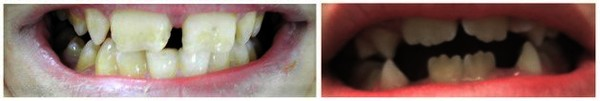
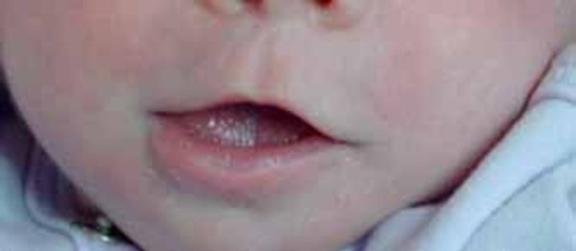
Fonksiyonel bozukluk, çiğneme ve konuşma problemlerini, diş bozunmalarını ve normal görme kaybını içerir. Hastalığın başlangıcı genellikle 14 ay ile 4 yaş arasındadır. Hastalık çocukluk çağında ilerler, daha sonra stabilize olur ve bazı durumlarda tedavi olmadan gerilemektedir.
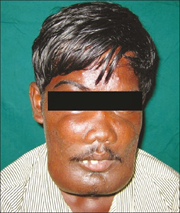
Görsel 1: Cherubism hastasında aşırı genişleyen çene kemiği
Cherubism
hastalarının çoğunda kromozom 4p16 üzerinde SH3BP2 geninde dominant
mutasyon bulunmaktadır. SH3BP2 tarafından kodlanan protein, kemik metabolizması
ve yeniden şekillendirilmesi için önemlidir. Cherubism’li insanların
yaklaşık %80’i SH3BP2 geninde bir mutasyona sahiptir. Vakaların
kalan %20’sinde durumun nedeni bilinmemektedir.
Belirti ve Semptomlar
Alt ve üst çene kemiğinin ağrısız, kist benzeri büyümelerle yer değiştirilmesiyle genişlemesidir. Bu büyümeler yanaklara şişmiş, yuvarlak bir görünüm verir ve genellikle normal diş gelişimine engel olur. Bazı insanlarda durum o kadar hafiftir ki fark edilmeyebilir, diğer vakalarda ise görme, nefes alma, konuşma ve yutma ile ilgili sorunlara neden olacak kadar şiddetlidir. Çenenin genişlemesi genellikle çocukluk dönemi boyunca devam eder ve ergenlik döneminde stabilize olur. Anormal büyümeler yavaş yavaş erken yetişkinlikte normal kemik ile değiştirilir. Sonuç olarak, etkilenen birçok yetişkin normal bir yüz görünümüne sahiptir. Bu durum başka hastalıklarla beraber görüldüğünde ise farklı fiziksel görünümler ortaya çıkmaktadır. Örneğin cherubism, ramon sendromu ile beraber görüldüğünde ise kısa boy, zihinsel engellilik ve diş etlerinin aşırı büyümesi gibi semptomlarla karşılaşılabilir. Bir nadir vaka olan noonan sendromu ile beraber görüldüğünde kısa boy ve kalp bozukluğu gibi durumlar da görülebilir.
Cherubism
görülme sıklığı tam olarak bilinmemektedir ve geniş
klinik spektrum nedeniyle belirlenmesi zordur. Literatürde dünya çapında çeşitli
etnik gruplarda yaklaşık 300 vaka
bildirilmiştir. Hastalık erkekleri ve kadınları eşit
derecede etkiler. Hastalığın yaklaşık
%50’si aileseldir ve diğerleri de
novo mutasyonlarıyla ilişkili
görünmektedir. Cherubism otozomal dominant bir özellik olarak kabul edilir,
ancak otozomal resesif iletimi düşündüren
belgeler de vardır. Hastalıktan etkilenen aileler için genetik danışmanlık
tavsiye edilir.
Kalıtım Paterni /
Deseni
Cherubism
otozomal dominant paternde kalıtsaldır. Bu, her hücrede sorumlu genin sadece
bir kopyasında bir değişikliğe
(mutasyon) sahip olmanın, durumun görülmesi için yeterli olduğu
anlamına gelir. Bazı durumlarda, hastalıktan etkilenen bir kişi
mutasyona uğramış geni yine
hastalıktan etkilenen bir ebeveynden devralır. Diğer
durumlarda mutasyon ailesinde hiç görülmemiş
bireylerde de ortaya çıkabilir. Buna de novo mutasyonu denir. De novo mutasyonunun
neden olduğu cherubism vakalarının oranı bilinmemektedir, çünkü bu
durumda değişken
ekspresivite ve azaltılmış penetrans
gözlenir.
Otozomal dominant bir duruma neden olan bir mutasyona sahip bir kişinin çocukları olduğunda, her çocuğun bu mutasyonu alma şansı %50’dir (2’de 1).
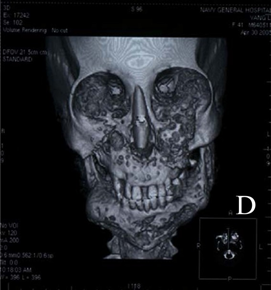
Görsel 3: 41 yaşındaki cherubism hastası bir kadına ait tomografi görüntüsünde alt çene kemiğinde fark edilebilir bir şekilde genişleme gözükmektedir.
Hastanın yaşı, aile
öyküsü, klinik bulgular, radyografik bulgular, histopatolojik bulgular, biyokimyasal
incelemeler ve moleküler analiz sonuçlarına bakılarak karar verilmektedir. Cherubism
genellikle kendini sınırlayan bir hastalık olduğundan
dolayı cerrahi tedavi her hastada gerekli olmayabilir. Uzun süreli takip ve
kontroller çoğu vakada yeterlidir. 1999’da agresif dev hücre
lezyonlarının tedavisi amacıyla interferon kullanımı gündeme gelmiştir. Cherubismin
aktif evresinde interferonun vasküler proliferasyon ve dev hücreler üzerine
etki göstererek faydalı olabileceği düşünülmektedir.
Fare deney çalışmalarında yüksek doz TNF-alfanın cherubism progresyonuna
neden olabileceği gösterilmiştir. Ancak
1 hastada TNF-alfa blokeri kullanımıyla (bifosfonat ile birlikte) klinik olarak
başarı elde edilememiştir.
Hastalıkla İlişkili Genler
Cherubismli insanların yaklaşık yüzde 80’inde Sh3bp2 geninde mutasyonlar tanımlanmıştır. Kalan vakaların çoğunda, durumun genetik nedeni bilinmemektedir. Sh3bp2 geni, kesin işlevi belirsiz olan bir protein yapmak için talimatlar sağlar. Protein, hücreler içinde kimyasal sinyallerin iletilmesinde, özellikle de eski kemik dokusunun yeni kemik dokusuyla ve bazı bağışıklık sistemi hücreleri ile değiştirilmesinde rol oynar. Sh3bp2 genindeki mutasyonlar, bu proteinin aşırı aktif bir versiyonunun üretimine yol açar. Sh3bp2 mutasyonlarının etkileri hala incelenmektedir, ancak araştırmacılar anormal proteinin kemik dokusunun ve bazı bağışıklık sistemi hücrelerinin korunmasıyla ilişkili hücrelerdeki kritik sinyal yollarını bozduğuna inanmaktadır. Aşırı aktif protein muhtemelen çene kemiklerinde iltihaplanmaya neden olur ve kemiğin yeniden şekillendirilmesi sırasında kemik dokusunu parçalayan hücreler olan osteoklastların üretimini tetikler. Bu kemik yiyen hücrelerin fazlalığı, üst ve alt çenelerde kemiğin tahrip edilmesine katkıda bulunur. Kemik kaybı ve inflamasyonun bir kombinasyonu, muhtemelen, kistin karakteristik büyümelerinin temelini oluşturur.
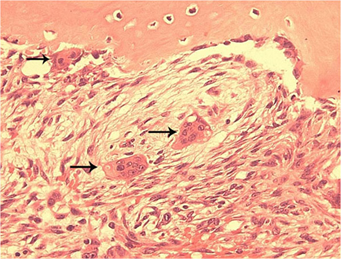
Görsel 4: Cherubismin tipik histopatolojisi. Bir cherubism lezyonundan histolojik bir kesit, kemiğin yakınında ve yumuşak fibröz stroma içinde çok çekirdekli osteoklast benzeri dev hücrelerin (oklar) tipik bulgularını göstermektedir.
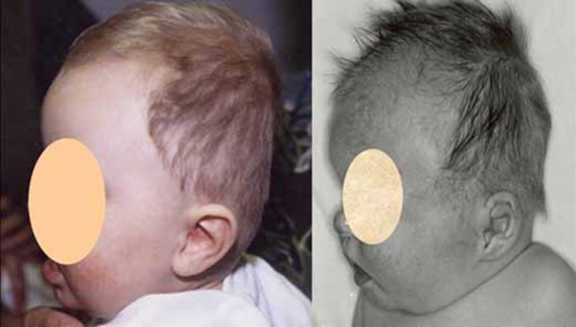
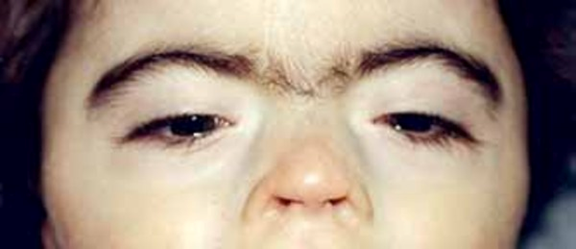
Saethre-Chotzen sendromu, bazı kafatası kemiklerinin (krainosinostoz) erken füzyonu ile tanımlanan genetik bir durumdur. Bu erken füzyon kafatasının normal şekilde büyümesini önler ve kafa ile yüzün şeklini ve simetrisini etkiler. Genellikle koni şeklinde bir kafa, asimetrik yüz, saç hizasının aşağıda olması ve sarkık göz kapakları (ptosis) şeklinde tanımlanan bir sendromik kraniyosinostoz şeklidir.
SCS’u hücre soyunun belirlenmesiden ve farklılaşmasından
sorumlu temel bHLH kopyalama faktörünü kodlamakla görevli TWIST1 (7p21) geninin
nokta mutasyonlara uğraması veya silinmesi nedeniyle gerçekleşir. Bu gendeki
mutasyonlara erken kafatası birleşmesine eden olur. Gen silmeleri genellikle
önemli nörokognitif gecikmelerle ilişkili olan daha ciddi fenotiplere neden
olur. FGFR3, FGFR2 and TCF12’deki mutasyonların, fenotip olarak SCS ile örtüşen
sinostoz koşullarına neden olduğu rapor edilmiştir.
Belirti ve Semptomlar
SCS’ nun belirti ve semptomları aynı aileden etkilenen
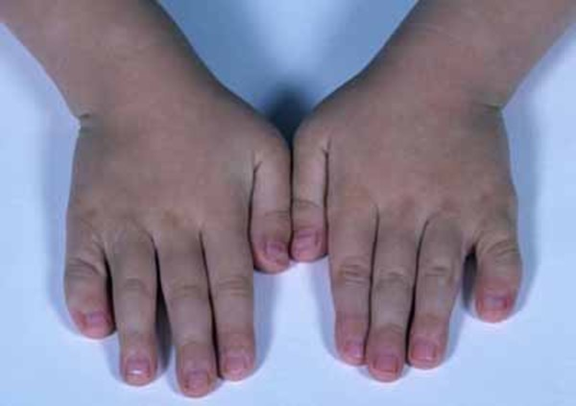
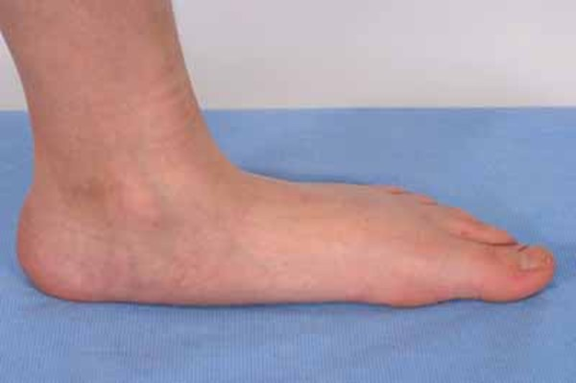
bireyler arasında bile geniç ölçüde çeşitlilik gösterir.Bu durum, ellerde ve
ayaklarda hafif anormalliklere neden olabilir örneğin her bir eldeki ikinci ve
üçüncü parmakların birleşmesi ve geniş veya kopyalanmış büyük ayak parmağı
gibi. Gecikmiş gelişme ve öğrenme zorluğu bildirilmiştir, ancak bu duruma sahip
çoğu kişi normal zekaya sahiptir. Çok yaygın olmayan belirti ve semptomlarından
bazılarıda kısa boyluluk, omurga kemiklerindeki anormallikler, işitme kaybı ve
kalp hasarıdır.
Genetik Görülme Sıklığı
Saethre-Chotzen sendromu tahmini 25.000 ila 50.000 kişide
bir görülme sıklığına sahiptir.
Kalıtım Paterni/Deseni
Bu durum otozomal dominant şekilde kalıtılır, bu durumda
da her hücredeki değiştirilmiş genin bir kopyası bozukluğa neden olur. Bazı
durumlarda, etkilenen kişi etkilenen bir ebeveynin mutasyonunu devralır. Diğer
durumlar, gendeki yeni mutasyonlardan kaynaklanabilir. Bu vakalar ailelerinde
herhangi bir bozukluk olmayanlarda görülür.
Bazı insanlar TWIST1 mutasyonlı oldukları halde SCS’unun
belirgin bir özelliğine sahip değillerdir. Bu insanlar hala bu gen mutasyonunun
çocuklarına geçme ve diğer belirtileri ve semptomları gösterme riski
altındadırlar.
Teşhis Yöntemleri ve Tedaviler
SCS tedavisi, kraniyofasiyal bir ekip tarafından genç
erişkinliğe kadar takip gerektirir. Genel olarak, hastalar intrakraniyal hacmi
arttırmak ve anormal bir kafa şeklini eski haline getirmek için yaşamın ilk
yılından bir kraniyoplasti (kafatasının bir kusurunun veya deformasyonunun
cerrahi onarımı) yaptırmalıdır.
Çocukluk çağında, solunum yolu tıkanıklığı ve
maloklüzyon tedavisi için orta yüz cerrahisi gerekli olabilir. Yarık
damaktakilerde, diğer malformasyonlar ve gerekli olan konuşma terapisi
bağlamında cerrahi kapatma yapılabilir. Yüz büyümesinin, işitme kaybının ve
psikomotor gelişimin rutin değerlendirmelerine ek olarak, şaşılık, ambliyopi
veya kronik papilödem (artmış ICP’yi gösterir) izlemek için düzenli
oftalmolojik muayenelere ihtiyaç vardır. Gelişimsel gecikme olan çocuklara
erken müdahale programları önerilmelidir.
Genel
Bilgi, Genetik Değişiklikler/Etken Faktörler
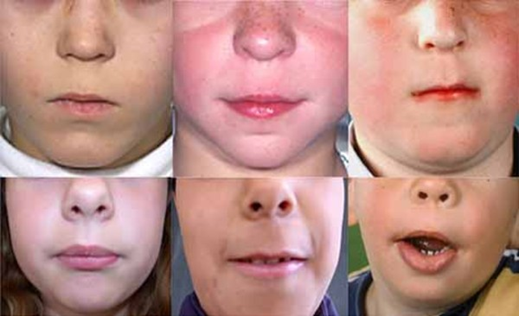
Pallister-Hall sendromu (PHS), vücudun birçok
bölümünün gelişimini etkileyen genetik bir hastalıktır. Ortak özellikler
arasında ekstra parmaklar ve / veya ayak parmakları (polidaktil), parmaklar
veya ayak parmakları arasında ekstra cilt (sindaktil), beyinde hipotalamik
hamartom denilen bir anormal büyüme ve iki üçlü epiglotis olarak bilinen hava
yolunun bir malformasyonu bulunur. Nadir durumlarda bifid epiglot, solunum
yetmezliğine yol açabilir. Çoğu durumda hipotalamik hamartom sorun yaratmasa
da, bazı durumlarda nöbetler, büyüme hormonu eksikliği, erken ergenlik veya
kortizol eksikliğine neden olabilecek birçok hormonun (panhipopitüitarizm)
eksikliği gibi nörolojik sorunlara neden olabilir. PHS’nin diğer semptomları
arasında deliksiz anüs, böbrek anomalileri, kalp defektleri, küçük genital
organlar, parmak eksikliği, tırnak problemleri, yarık damak, bifid uvula ve
gelişim gecikmesi ve davranış problemleri sayılabilir. GLI3 genindeki
mutasyonlar Pallister-Hall sendromuna neden olur. Bu gen, genlerin belirli
hücrelerde açılıp kapatılmayacağını düzenleyen bir işlem olan gen ekspresyonunu
kontrol eden bir protein yapmak için talimatlar sağlar. Gelişim sırasında
belirli zamanlarda belirli genlerle etkileşime girerek, GLI3 proteini doğumdan
önce birçok organ ve dokunun normal şekillenmesinde (desenlenmesinde) rol
oynar.
Pallister-Hall sendromuna neden olan
mutasyonlar tipik olarak GLI3 proteininin anormal derecede kısa bir
versiyonunun üretilmesine yol açar. Hedef genleri açıp kapatabilen normal GLI3
proteininin aksine, kısa protein sadece hedef genleri kapatabilir
(bastırabilir). Araştırmacılar, protein işlevindeki bu değişikliğin erken
gelişimi nasıl etkilediğini belirlemek için çalışmaktadır. GLI3 mutasyonlarının
polidaktil, hipotalamik hamartom ve Pallister-Hall sendromunun diğer
özelliklerine neden olabileceği kesin değildir.
Belirti ve
Semptomlar
PHS’li çoğu hasta doğumda üçüncü veya dördüncü
dereceden iskelet polidaktili veya postaksiyal polidaktili görülürken,
beraberinde her ikisine de kütanöz sindaktili ve tırnak displazisi eşlik
edebilir. Yüz özellikleri kısa burun, düz burun köprüsü ve alçak ayarlanmış,
arka acılı kulakları içerebilir. Bazı hastalarda yarık damak, yarık uvula ve
multipl bukal frenula bildirilmiştir. Asemptomatik bir bifid epiglotis
neredeyse patognomoniktir, ancak bazı hastalarda potansiyel olarak ölümcül
solunum yetmezliğine yol açan daha ciddi posterior laringeal yarıklar görülür.
Hipotalamik hamartom genellikle asemptomatiktir, ancak panhipopituitarizm ile
ilişkili olabilir. Akut primer adrenal yetmezlik ciddi vakaların yani sıra,
daha hafif adrenal yetmezlik formlarında da görülebilir. Erken ergenlik bazı
durumlarda kendini gösterir. Nörolojik tutulum, gelastik epilepsiyi (yüz
ekşitmeden, gülüşmekten veya kahkaha olarak ortaya çıkan nöbetler) veya diğer
nöbet tiplerini içerebilir. Vajinal atrezi veya hidrometrokolpos, microphallus
veya kriptorşidizm de dahil olmak üzere böbrek agenezisi veya displazisi ile
diğer genitoüriner anomaliler bildirilmiştir. Diğer bulgular arasında
intrauterin gelişme geriliği, anormal akciğer lobasyonu, mezomelik kısalması
ile genelleşmiş iskelet displazisi ve uzuvların radyal yaylanması, imperforat
anüs ve konjenital kalp defektleri sayılabilir.
Genetik Görülme Sıklığı
Bu durum çok nadirdir; prevalansı
bilinmemektedir. Bugüne kadar yaklaşık 100 hasta bildirilmiştir.
Kalıtım
Paterni/Deseni
Bu durum otozomal dominant paternde kalıtsaldır, bu da her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir. Bazı durumlarda, etkilenen bir kişi, etkilenen bir ebeveynden GLI3 geninde bir mutasyon devralır. Diğer vakalar, gendeki yeni mutasyonlardan kaynaklanır ve ailelerinde hastalık öyküsü olmayan kişilerde ortaya çıkar.
Iafolla ve diğerleri (1989) manyetik rezonans
görüntülemenin en değerli tanı aracı olduğuna işaret etmişlerdir; BT
taramasının tümörü kaçırdığı bildirildi.
Pallister-Hall sendromu üzerine uluslararası
bir atölye çalışması (Biesecker ve diğerleri, 1996) bu varlık için minimal tanı
kriterleri geliştirmiştir. Bir ailedeki indeks olgusunda, tanı kriterlerini
karşılamak için hem hipotalamik hamartom hem de merkezi polidaktili olmalıdır.
İndeks olgunun birinci derece akrabalarında hipotalamik hamartom veya
polidaktili (merkezi veya postaksiyal) bulunmalı ve otozomal dominant paternde
veya gonadal mozaikliği ile uyumlu bir şekilde kalıtım gösterilmelidir. Şüpheli
olguların klinik değerlendirmesi için öneriler sunuldu. Biesecker ve diğerleri
(1996), hipotalamik hamartomun PHS’ye özgü olmadığı sonucuna varmıştır.
Hastalıkla İlişkili Genler
Pallister-Hall sendromuna GLI3 genindeki
mutasyonlar neden olur. Kalıtım otozomal dominanttır, ancak vakaların yaklaşık
dörtte birinde Pallister Hall sendromu yeni (de novo) bir mutasyondan
kaynaklanır
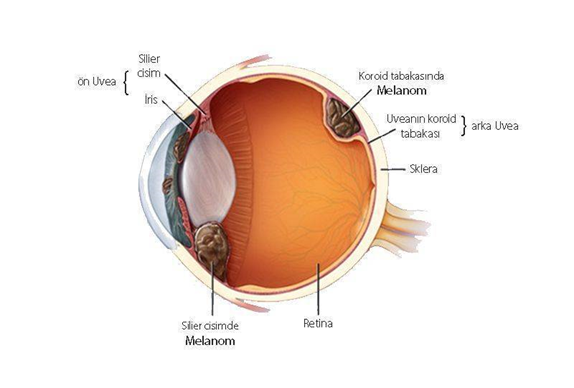
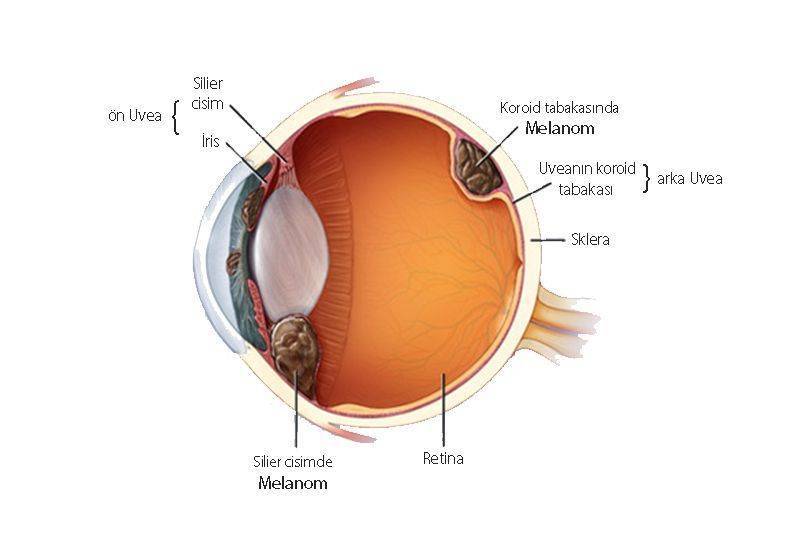
İris melanom, Göz melanomu, nadir kanserler sınıfında değerlendirilir. İris melanom vakaların % 90’ında koroidden ve vakaların diğer % 10’unda iris ve siliyer cisimden kaynaklanan ve klinik olarak görsel semptomlarla (bulanık görme, fotopsi, yüzer ve görme alanı azaltma), görünür bir kitle ve ağrı ile baş gösterir. Tüm hastaların yaklaşık yarısında ölümcül metastatik hastalık görülür ve karaciğer en sık metastaz bölgesidir.
Yetişkinlerde en sık görülen primer göz içi malignitesi olan iris melanom, iris, siliyer cisim ve koroid içindeki melanositlerden kaynaklanır. Melanom her yaşta gelişebilir, ancak en sık ellili yaşlarında yetmişli yaşlarda görülür ve gençler ve genç yetişkinlerde daha yaygın hale gelir.
İris melanom tanısındaki gecikme çalışmaları, bu lezyonların% 28-37’sinin ilk muayenede tespit edilmediğini göstermektedir. Tanı ve tedavideki ilerlemelere rağmen, iris melanom yaşamı tehdit eden bir malignite olmaya devam etmektedir ve uzun süreli takipte hastaların yaklaşık yarısında sistemik metastaz görülmektedir.
İris melanomun lokal ve sistemik tedavisinde tanı ve ilerlemeler, son birkaç on yılda transpupiller termoterapi ve radyoterapiyi içeren enükleasyondan göz koruyucu tedavi yöntemlerine geçişe neden olmuştur.
Çoğu melanom vakası sporadiktir, bu da ailelerinde rahatsızlık öyküsü
olmayan kişilerde ortaya çıktığı anlamına gelir.
İris melanom adı verilen bir tür göz kanseri BAP1 tümör predispozisyon sendromunda en sık görülen kanserli tümör hastalığıdır.
Melanom, melanositlerden kaynaklanan malign bir tümördür ve deriden (% 91), göz ve göz çevresindeki dokulardan (% 5) veya mukozadan (% 1) kaynaklanabilir. Hastaların% 2’sinde kaynak tanımlanamaz. İris (% 85), göz kapağı / orbita (% 10) ve konjonktivada (% 5) oftalmik melanomlar ortaya çıkabilir.
Melanom ile İlişkili Melanositik Lezyonlar
· · Koroid nevüsü: Koroidal nevüslar 30 yaşın üzerindeki bireylerin% 3’ünde bulunur ve çalışmalar, malign dönüşüm oranlarının 8,845’te 4.300’de 1’den 1’e değişebileceğini göstermektedir.
· Oküler / Okülodermal melanositoz: Oküler veya okülodermal melanositoz, episklera, uvea ve cildin hiperpigmentasyonu ile karakterize edilen ve siyah, İspanyol ve Asya popülasyonlarında daha yaygın olan bir durumdur. Beyazlardaki prevalansı% 0.04’tür ve 400 vakadan 1’inde iris melanom gelişir.
· Kutanöz nevüs: Vaka-kontrol çalışmaları, kutanöz nevusların uveal melanom için bir risk faktörü olabileceğini ve displastik nevüs sendromlu hastaların iris melanom insidansının daha yüksek olduğunu göstermiştir. Bu, iris melanom hastalarında dermatolojik değerlendirme ihtiyacını vurgulamaktadır.
· Ailesel iris melanom: Son zamanlarda, germ hattı BAP1 mutasyonu olan bazı hastalarda otozomal dominant kalıtsal kanser sendromu tanımlanmıştır. Bu mutasyona sahip hastalar, daha yüksek iris melanom, kutanöz melanom, atipik Spitz tümörleri, mezotelyoma, menenjiyom, akciğer adenokarsinomu ve diğer birçok kanser tipine sahiptir.
İris melanomdaki genetik değişiklikler karyotipleme, tek nükleotid polimorfizmi, floresan in situ hibridizasyon, mikrosatellit analizi ve deoksiribonükleik asit (DNA) seviyesinde karşılaştırmalı genomik hibridizasyon ve ribonükleik asit (GEP) ile ribonükleik asit (GEP) ile araştırılır. RNA) düzeyindedir. Birçok genin haberci RNA (mRNA) ekspresyonunu değerlendiren RNA bazlı GEP çalışmalarında, uveal melanom iki gruba ayrılabilir: düşük metastaz riski olan hastalar (sınıf 1) ve yüksek metastaz riski olan hastalar (sınıf 2).
Uveal melanomda kötü prognoz ile ilişkili olduğu bildirilen ilk
kromozomal değişiklik, kromozom 3 monozomisidir.
İris melanoma neyin sebep olduğu tam olarak bilinmemektedir, fakat melanom da diğer tüm kanserler gibi hücrelerdeki DNA hasarları sonucu oluşmaktadır. DNA hasarlarının birikmesi, hücrenin programlı hücre ölümüne (apoptoz) gitmesini engeller ve kontrolsüz çoğalma başlar. Mavi ya da yeşil gözlü insanlar, daha fazla gözde melanom geliştirme riskine sahiptir. Açık tenli olmak, ırk olarak açık renkli ten rengine sahip kişiler diğer ırklara oranla daha fazla risk altındadır. Ultraviyole (UV) ışınlarına maruz kalmak. UV ışınlarına güneş ya da solaryum gibi yollarla maruz kalan gözlerde melanom riski artmaktadır.
Belirti ve Semptomlar
Her ne kadar iris melanom genellikle herhangi bir semptom göstermezse de, bu tür kanserli bazı kişilerde bulanık görme vardır; görüşlerinde küçük, hareketli noktalar (yüzer) veya yanıp sönen ışık; baş ağrısı; veya gözün üzerinde gözle görülür bir karanlık nokta olabilir. Siliyer cisim malign melanomunun özel anatomik yeri nedeni ile erken evrede tanı ve tedavisi zordur. Genellikle çok genişlediklerinde, görme kaybına ya da metastatik hastalığa neden olduklarında teşhis edilebilirler.
İris melanoma’da, Tanı esas olarak tümörün biyomikroskopi ve dolaylı oftalmoskopi ile klinik muayenesine dayanır ve ultrasonografi, fundus floresein anjiyografi ve optik koherens tomografi gibi tanı teknikleri ile doğrulanır. Dilate fundus muayenesinde pigmente kubbe şeklindeki kitlenin klasik görünümü tespit edildiğinde posterior iris melanomların klinik tanısı konabilir. İris melanomlar klasik olarak A-tarama ultrasonografisinde ve B-tarama ultrasonografisinde düşük ila orta yansıtma gösterirler, tümör hiperekoik, akustik olarak içi boş bir göz içi kütlesi olarak görülür. Şüpheli pigmentli bir lezyonun yönetimi, büyüme, 2 mm’den büyük kalınlık, subretinal sıvının varlığı, semptomlar ve turuncu pigment, optik diskin 3 mm dahilindeki marj, ve halo ve drusen yokluğu.
Malign melanomu ailesel geçiş gösteren ve göstermeyen olarak
sınıflandırdığımızda, yaklaşık %10 kadarının kalıtsal olduğu ortaya
çıkmaktadır. Genom analiz çalışmaları ile hangi kromozom ve gen bozukluklarının
melanoma neden olduğunu ve bu hatalara sahip bireylerin melanoma yakalanma
riskleri öngörebilmektedir.
İris melanom’da prevalansın milyon erkek başına 4.9 ve milyon kadın başına 3.7 olduğu tahmin edilmektedir. BAP1 tümör predispozisyon sendromu nadir bir durumdur; yaygınlığı bilinmemektedir, hastalıklı 70’den fazla aile tıbbi literatürde tanımlanmıştır.
Kalıtım Paterni / Deseni
BAP1 geninde mutasyonu olan kişiler, tümör oluşumu riskini arttırır. Gen mutasyonu
olan tüm insanlar bir tümör geliştirmeyecektir.
Uveal melanom
gelişimini içerebilen ve kromozom 3p21 üzerinde BAP1 genindeki
( 603089 ) germ hattı mutasyonundan kaynaklanan
bir tümör yatkınlık sendromu için ayrıca bkz . 614327.
Teşhis Yöntemleri ve Tedavileri
Teşhis
Yetişkinlerde en sık görülen primer göz içi malignitesi olan iris melanom, iris, siliyer cisim ve koroid içindeki melanositlerden kaynaklanır. Tanı esas olarak tümörün biyomikroskopi ve dolaylı oftalmoskopi ile klinik muayenesine dayanır ve ultrasonografi, fundus floresein anjiyografi ve optik koherens tomografi gibi tanı teknikleri ile doğrulanır. Dilate fundus muayenesinde pigmente kubbe şeklindeki kitlenin klasik görünümü tespit edildiğinde posterior iris melanomların klinik tanısı konabilir. Uveal melanomlar klasik olarak A-tarama ultrasonografisinde ve B-tarama ultrasonografisinde düşük ila orta yansıtma gösterirler, tümör hiperekoik, akustik olarak içi boş bir göz içi kütlesi olarak görülür. Prognoz, tedaviden önce tümörün ince iğne aspirasyon biyopsisinin genetik profillenmesi ile en doğru şekilde tahmin edilebilir.
Ayırıcı tanı
İris melanom iris, siliyer cisim ve koroid melanomuna ayrılır ve bu alt tiplerin ayırıcı tanısında bazı lezyonlar düşünülmelidir. İris melanomu dışında şüpheli bir iris lezyonunun olası tanıları arasında iris nevus, iris pigment epitel kisti, iris stromal kist, irisin metastatik tümörü, melanositoma, iris atrofi ve Cogan-Reese sendromu bulunur. Melanomun yanı sıra siliyer cisim tümörlerinin ayırıcı tanısında stafiloma, medulloepithelioma ve leiomyoma bulunmalıdır. Uveal melanomların çoğu, çeşitli lezyonlarla simüle edilebilen koroid melanomudur. Bunlar arasında koroid tümörleri, özellikle koroid nevüsü, metastatik tümörler, koroid hemanjiyomu ve osteoma; AMD gibi hemorajik durumlar ve hemorajik koroid dekolmanı; konjenital retinal pigment epitel hipertrofisi ve retinal pigment epitel adenokarsinomu gibi retinal tümörler; ve posterior sklerit gibi enflamatuar lezyonlar.
İris melanomlar, iç tümör dolaşımının yanı sıra koroid dolaşımına sahiptir. Tanıyı doğrulamak için bu çift dolaşım paterninin gözlemlenmesi veya tümöral vaskülatürden sızıntı bazen gereklidir. Bu özellikleri görselleştirebilen FFA, diğer lezyonlardan ayırıcı tanı sırasında önemli bir tekniktir. FFA ayrıca, radyasyon retinopatisi ve radyasyon makülopatisi gibi brakiterapi sonrası ortaya çıkan komplikasyonların saptanmasında ve izlenmesinde de kullanılır.
Bilgisayarlı tomografi (BT) ve manyetik rezonans görüntüleme
(MRI), klinik muayene ile tümör görselleştirmesi, katarakt, vitreus kanaması
veya retina dekolmanı gibi ortam opasiteleri olan hastalarda olduğu gibi bir
zorluk oluşturduğunda kullanılabilir. Özellikle tek taraflı kataraktı olan
hastalar, siliyer cisim melanomunun merceğe uygulanan basınçla tek taraflı veya
asimetrik katarakta neden olabileceğini akılda tutarak uveal melanom açısından
dikkatle değerlendirilmelidir.
Göz içi tümörleri birkaç yöntemle biyopsi yapılabilir. Ön
segment tümörleri sulu mizah örneklemesi, insizyonel veya eksizyonel biyopsi
ile değerlendirilebilir. Posterior segment intraoküler tümörleri
değerlendirmek için FNAB (transskleral, transvitreal veya transcameral),
vitrektomi biyopsisi, insizyonel veya eksizyonel biyopsi (endoreseksiyon veya
transskleral rezeksiyon) yapılabilir.
Tedavi
İris melanom tedavisi tanı sırasında başlatılır ve sadece göz içi tümörü ile sınırlı değildir. İris melanomun yönetimi ayrıca klinik uygulamada şu anda kullanılan prognostik faktörlerin değerlendirilmesini ve gerektiğinde sistemik hastalığı hedefleyen adjuvan tedavilerin planlanmasını; tedavi sonrası nüks ve tedaviye bağlı oküler yan etkilerin izlenmesi ve kontrolü; uygun tedavi seçeneklerini kullanarak görsel fonksiyon değerlendirmesi ve görsel rehabilitasyon; metastaz riski için rutin sistemik değerlendirme; ve psikiyatrik değerlendirme. Bu adımlardan herhangi birinin ihmal edilmesi, tümörün başarılı tedavisine rağmen, ölümle sonuçlanan tedavi başarısızlığına yol açabilir.
Şu anda kabul edilen ve klinik olarak uygulanan tümör yönetimi
anlayışı, prognostik faktörlerin uygun bir değerlendirmesi ile başlar, bundan
sonra hem tümörü kontrol etmek hem de sağlıklı dokulara olan etkiyi en aza
indirmek için bu faktörler göz önünde bulundurularak bir veya daha fazla tedavi
seçilir. Hangi tedavi seçeneklerinin uygun ve uygulanabilir olduğuna
ilişkin kararlar tümör boyutu, yeri ve genişlemesine göre yapılır ve hastanın
tercih ve beklentileri dikkate alınır.
Sistemik metastazı olmayan iris melanom hastaları için iki ana tedavi seçeneği göz koruyucu tedaviler ve enükleasyondur. Çalışmalar, tedavi yöntemlerindeki gelişmelere ve son 30 yılda göz koruyucu tedavilere yönelik artan eğilime rağmen, hayatta kalma oranlarının sabit kaldığını göstermiştir. Bu, gözün başarılı lokal tedavisinin sağkalımı etkilemediğini gösterir. Bu nedenle, metastaz riski olan hastaların belirlenmesi ve lokal tedaviye ek olarak adjuvan tedavilere yönlendirilmesi çok önemlidir.
Bugüne kadar,
küçük, pigmentli bir koroid tümörü ile karşılaşıldığında geleneksel yaklaşım,
renkli fundus fotoğrafçılığı bulguları büyümeyi gösterene kadar lezyonu
izlemektir. Küçük tümörler literatürde malign transformasyonu gösteren faktörler
bağlamında değerlendirilmeli ve hastaları tedavide yer alan yararlar ve riskler
hakkında tam olarak bilgilendirildikten sonra kararlar alınmalıdır. Bununla
birlikte, bir tümörün tedavi gerektiren bir boyuta ulaşmadan önce metastatik
hale gelip gelmeyeceğini bilmek imkansız olduğundan, tedaviyi geciktirmek
metastatik yayılım ile sonuçlanabilir. Öte yandan, küçük melanomların %
30-40’ının optik disk ve makulaya yakın olduğu düşünüldüğünde, tüm şüpheli
koroid tümörlerinin tedavi edilmesi gereksiz oküler morbidite ve görme kaybına
neden olacaktır.
Göz Koruyucu Tedaviler Fotokoagülasyon
Terapi Fotokoagülasyon
Fotokoagülasyon
geçmişte küçük koroid melanomunu tedavi etmek için sıklıkla kullanıldı, önce
ksenon ark ve daha sonra argon lazer fotokoagülasyonu. Ksenon ark
fotokoagülasyonu ile üstün tümör kontrolüne rağmen, argon lazer daha az
komplikasyona neden olur. Bugün, 3 mm’den daha kalın ve foveadan 3 mm’den daha
fazla bulunan küçük tümörler, transpupiller termoterapi (TTT) ile tedavi
edilmektedir.
Transpupiller Termoterapi
TTT, küçük ve orta boy melanomları (tümör kalınlığı 4 mm’den az) tedavi etmek için kullanılan diyot lazer tabanlı bir yöntemdir. İris melanom nedeniyle TTT uygulanan hastalar, görsel prognoz iyi olmasına rağmen, uzun süreli yüksek metastatik riskli nüks olasılığı olduğunu akılda tutarak dikkatle seçilmelidir.
Radyoterapi
Radyoterapi şu anda iris melanom, özellikle posterior uveal melanom için en yaygın tedavidir. Klinik uygulamada radyoterapi, radyoaktif plak, harici ışın radyoterapisi veya SRT şeklinde uygulanabilir.
Brakiterapi,
tümörün yüzeyine veya iç kısmına radyoaktif bir kaynak (radyoizotop)
uygulanması yoluyla bir tümörün doğrudan ışınlanmasıdır.
Dış Işın Tedavisi
Dış ışın
tedavisi, bir tümörün proton ve helyum iyon ışınları gibi yüklü parçacıklarla
veya stereotaktik yöntemlerle ışınlanmasıdır.
Proton Işın Tedavisi
Brakiterapi ve fraksiyone SRT’den farklı olarak, proton ışını tedavisi tüm tümöre homojen bir radyasyon dozu verir ve Bragg etkisi nedeniyle radyasyon hedefin kenarının hemen ötesine dağılır. Bu, bitişik normal dokuyu koruyarak tümöre yüksek dozda radyasyon verilmesini sağlar; bununla birlikte, ışının vücuda girdiği ve tümörü hedeflediği yoldaki dokular da yüksek dozda radyasyon alır. Teorik olarak, tüm iris melanomlar proton ışını tedavisi ile tedavi edilebilir, ancak büyük melanomlar için görsel prognoz ve göz koruma oranları düşük kalır.
Stereotaktik Radyoterapi
SRT, bir
tümörün bir foton ışını ile ışınlanmasıdır. SRT’de radyasyon tek bir doz
olarak verilirken, kesirli SRT’de toplam doz daha küçük eşit dozlar olarak
verilir. Stereotaktik bir foton ışını ile tedavinin bir avantajı, tümörün
yerini belirlemek için herhangi bir cerrahi prosedür gerekmemesi, tümör
sınırlarının MRG ve BT ile belirlenmesidir.
Gama Bıçağı
İlk olarak beyin tümörlerinin tedavisinde uygulanan Gamma Knife o zamandan beri iris melanomları başarılı sonuçlarla tedavi etmek için kullanılmıştır.
CyberKnife
CyberKnife radyocerrahi ilk kez beyin cerrahisinde kullanıldı ve şu anda iris melanomu tedavi etmek için kullanılıyor.
Doğrusal hızlandırıcı: Doğrusal hızlandırıcı, uveal melanomu stereotaktik hipofraksiyonlu radyoterapi ile tedavi etmek için kullanılır. Bu yaklaşımın avantajları, tümöre bitişik sağlıklı dokulara daha az radyasyon maruziyeti ve uzun süreli etkilerin önlenmesidir.
Cerrahlık
Enükleasyon, Yoklama, Lokal Rezeksiyon
Enükleasyon geçmişte en yaygın tedavi seçeneği olmasına rağmen, şu anda optik sinirleri çevreleyen koroidal melanomlu veya sunumu olan büyük iris melanomlu hastalar (8 mm’den büyük tümör kalınlığı) gibi en kötü görsel prognoza sahip vakalar için ayrılmıştır.
Lokal rezeksiyon,
gözü koruyan ve daha da önemlisi ayrıntılı bir histopatolojik ve sitogenetik
analize izin veren koroid melanom hastaları için alternatif bir tedavi
seçeneğidir.
Adjuvan Tedaviler
İris melanomun tanı ve tedavisinde kaydedilen ilerlemelere rağmen, metastatik hastalık nedeniyle genel mortalite yüksek olmaya devam etmektedir. Bu nedenle, metastaz riski yüksek olan hastaları tanımlamak son derece önemlidir. Adjuvan tedaviler şu anda klinik olarak tanımlanmış makrometastazları hedef alan radyoterapi ve sistemik tedaviden oluşmaktadır. Sistemik tedavi seçenekleri arasında kemoterapi, immünoterapi, hormon tedavisi, biyolojik tedavi ve hedefe yönelik tedavi yer alır. Diğer tümörlerin aksine, iris melanom ile ilgili hala çok az çalışma vardır.
Sonuç olarak, iris melanom tanısı ve lokalize hastalık tedavisinde ilerlemelere rağmen, hastaların yarısına kadar ölümcül metastatik hastalık gelişecektir. Tümörün moleküler manzarasını anlama ve GNAQ / GNA11, BAP1, EIF1AX, SF3B1 mutasyonları ve epigenetik mekanizmaları içeren yolları hedefleyen tedavilerin geliştirilmesindeki ilerlemelerle, yakın gelecekte mikrometastazların ilerlemesini önlemek mümkün olabilir.
Hastalıkla İlişkili Genler
RAS ve B-RAF gen mutasyonları yoluyla aktivasyon, kutanöz
melanomda yaygındır. Her ne kadar uveal melanomda MAPK yolu
aktivasyonu bildirilmiş olsa da, B-RAF veya RAS mutasyonları nadirdir.
3p21 kromozomu üzerinde bulunan BRCA-1 ile ilişkili protein-1
(BAP1) tümör baskılayıcı geni, kanser hücrelerinde tümör baskılama
aktivitesinden sorumlu enzimler arasında yer alan ubikuitin karboksi terminal
hidrolaz enzimini kodlar ve bazı proteinlerin aktivitesini düzenler
deubiquitination yoluyla. Örneğin histon H2A, belirli genlerin
ekspresyonunu düzenler; BAP1 bölgesinin deubikuitinasyonu, tümör baskılama
fonksiyonunda kritik bir adımdır.
Daha önce tarif edilen mutasyonların aksine, ekleme faktörü 3b alt birim 1 (SF3B1) gen mutasyonları iris melanomlarda % 19 oranında bulunmuştur ve iyi prognozla ilişkilidir.
İyi prognozla ilişkili bir başka mutasyon, iris melanomların% 24’ünde bildirilen ökaryotik çeviri başlatma faktörü 1A (EIF1AX) gen mutasyonudur.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
KGB
sendromu ilk 1975 yılında tanımlanıyor. Bu sendromun ismi ise Dr Optiz
tarafından bu sendromu takip ettiği ailelerin soyisim ilk harflerinden
gelmektedir.(1)
KBG
sendromu, üst merkezi kesici dişlerin makrodontisi, ayırt edici kraniyofasiyal
bulgular, kısa boy, iskelet anomalileri ve global gelişimsel gecikmeyi,
nöbetleri ve zihinsel engeli içeren nörolojik tutulumla karakterizedir. KBG sendromunun yetersiz teşhis edilmesinin
muhtemel olduğunu, çünkü zihinsel engellilik de dahil olmak üzere birçok
özelliğin ılımlı olduğunu ve hiçbir özelliğin teşhis için önkoşul
olmadığını belirtiliyor (2).
Belirti ve Semptomlar
KBG sendromunun karakteristik
bir özelliği alışılmadık derecede büyük üst ön dişlerdir (makrodonti). Diğer
belirgin yüz özellikleri arasında geniş, kısa bir kafatası ( brakisefali),
bir üçgen yüz şekli, geniş aralıklı gözler (hipertelorizm), ortada birlikte
büyüyebilecek geniş kaşlar (synophrys), belirgin bir burun köprüsü, burun ve
üst dudak arasında uzun bir boşluk (uzun filtrum) ve ince üst dudak
bulunur.
KBG sendromu olan
kişilerde yaygın bir iskelet anomalisi olarak kemiklerin mineralizasyonu
yavaşlamıştır (gecikmiş kemik yaşı); örneğin, etkilenen 3 yaşındaki bir
çocuğun 2 yaşında tipik bir kemiği olabilir. Ayrıca, etkilenen bireylerin
omurgalarında (omur) ve kaburga kemiklerinde anormallikler
olabilir. Ayrıca, olağandışı kısa ya da kavisli beşinci (pembemsi)
parmaklar ( brakidaktili ) dahil olmak üzere, ellerin veya ayakların
kemiklerinde anormallikler olabilir. (klinodaktili, düz ayaklar ( pes
planus)).
KBG sendromunda zihinsel
ve hareket kabiliyetlerinin gelişimi de gecikmektedir. Etkilenen
bireylerin çoğu konuşmayı ve yürümeyi normalden daha geç öğrenir ve zihinsel
yetersizliği hafif ila orta şiddettedir. Bu hastalığı olan çoğu insan,
hiperaktivite gibi davranışsal veya duygusal problemlere
sahiptir; anksiyete; veya bozulmuş iletişim ve sosyal
etkileşimlerle karakterize otizm spektrum bozukluğu da ayrıca incelenmelidir.
KBG sendromunun daha az
yaygın özellikleri ise işitme kaybı, nöbetler ve kalp kusurlarını içerir.
Tekin vd. (2004)
tarafından bildirilen Anadolu’dan
bir ailede; baba ve 2 oğlu KBG sendromu tanısı aldı ve otozomal dominant
kalıtım olduğu genetik testler ile doğrulandı.(3) Fakat yine 2004 yılında
yayınlanan bir başka makalede ise Maegawa ve diğ. (2004) hafif
derecede etkilenen bir anneyi ve ağır şekilde etkilenen oğlunu
bildirmiştir. Erkeklerin kadınlardan daha ciddi bir şekilde etkilendiği
önceki raporlara dayanarak (örneğin, Parloir ve diğerleri, 1977 (4) ), bazı durumlarda kalıtımın X’e bağlı
olabileceğini öne sürülmüştür (5).
Teşhis Yöntemleri ve Tedaviler
Teşhis;Tam
bir klinik değerlendirme, ayrıntılı bir hasta ve aile öyküsü ve karakteristik fiziksel
bulguların tanımlanmasından sonra KBG sendromu tanısından
şüphelenilebilir. Teşhis ayrıca gen paneli analizi veya zihinsel
yetersizliğin çoklu genetik nedenlerinin aynı anda araştırıldığı yeni nesil
dizilim teknikleri ile de yapılabilir.
Tedavi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını
gerektirebilir. Çocuk doktorları, ortopedistler, ortopedi cerrahları,
nörologlar, fizyoterapistler, konuşma terapistleri, ortodontistler ve diğer sağlık
uzmanlarının etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir
şekilde planlaması gerekebilir.
Genetik danışmanlık, etkilenen bireyler ve aileleri için
önerilmektedir. Tüm aileye psikososyal destek de faydalı olabilir.
Ortopedik cerrahi, etkilenen kişilerin kalça ve omurga
anormalliklerini düzeltmek için özellikle yardımcı olabilir. İşitme
cihazları, konuşma terapisi ve kapsamlı diş bakımı da faydalı olabilir.
Hastalıkla İlişkili Genler
KGB sendromuna ANKRD11 genindeki bir
değişiklik(mutasyon) veya ANKRD11 genini içeren 16q kromozomundan
genetik materyal kaybı neden olur. Genler, vücudun birçok fonksiyonunda kritik
bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir
genin mutasyonu meydana geldiğinde, protein ürünü hatalı, yetersiz veya eksik
olabilir. Belirli bir proteinin fonksiyonlarına bağlı olarak, bu vücudun
birçok organ sistemini etkileyebilir.
ANKRD11 gen sinir hücreleri
(nöronlar) aktif olan bir proteini oluşturmak için talimatlar içerir. Bu
proteinin tam rolü tam olarak anlaşılmamıştır. ANKRD11 değiştirildiğinde
veya kaybolduğunda, bireyler bu proteinin yeterince işlevsel kopyalarını
üretemezler. ANKRD11 geninin protein
ürününün düşük seviyelerinin KBG sendromu semptomlarına neden olmasına karar
vermek için daha fazla araştırma gereklidir .
KBG sendromu otozomal dominant paternde kalıtsaldır(3). Çoğu
genetik hastalık, biri babadan diğeri de anneden alınan bir genin iki
kopyasının durumuna göre belirlenir. Baskın genetik bozukluklar, belirli
bir hastalığa neden olmak için değiştirilmiş veya eksik bir genin sadece bir
kopyasının gerekli olduğu durumlarda ortaya çıkar. Etkilenen gen, her iki
ebeveynden miras alınabilir veya etkilenen bireyde yeni, kendiliğinden bir
mutasyonun (gen değişimi) sonucu olabilir. Buna “de novo” değişikliği
denebilir. Değiştirilen geni veya eksik kromozom segmentini etkilenen bir
ebeveynden yavrulara geçirme riski her hamilelik için% 50’dir. Risk
erkeklerde ve kadınlarda aynıdır.
Hastalığın Diğer İsimleri
Diğer isimleri
diyemesek de araştırmalarda şu şekilde de karşımıza çıkabilir:
Makrodonti,
zihinsel gerilik, karakteristik fasiyes, kısa boy ve iskelet anomalileri
boy
kısalığı-karakteristik fasiyes-zeka geriliği-makrodonti-iskelet
anomalileri sendromu
kısa boy,
karakteristik fasiyes, makrodonti, zeka geriliği ve iskelet anomalileri
Kaynakça
1) Herrmann, J., Pallister, P. D., Tiddy, W., Opitz, J. M. The KBG syndrome–a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Art. Ser. XI(5): 7-18, 1975.
2) Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., Bademci, G., Agolini, E., Guo, S., Konuk, B., Kavaz, A., Blanton, S., Digilio, M. C., Dallapiccola, B., Young, J., Zuchner, S., Tekin, M. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 89: 289-294, 2011.
3) Tekin, M., Kavaz, A., Berberoglu, M., Fitoz, S., Ekim, M., Ocal, G., Akar, N. The KBG syndrome: confirmation of autosomal dominant inheritance and further delineation of the phenotype. Am. J. Med. Genet. 130A: 284-287, 2004.
4) Parloir, C., Fryns, J. P., Deroover, J., Lebas, E., Goffaux, P., van den Berghe, H. Short stature, craniofacial dysmorphism and dento-skeletal abnormalities in a large kindred: a variant of KBG syndrome or a new mental retardation syndrome. Clin. Genet. 12: 263-266, 1977.
5) Maegawa, G. H. B., Leite, J. C. L., Felix, T. M., da Silveira, H. L. D., da Silveira, H. E. Clinical variability in KBG syndrome: report of three unrelated families. Am. J. Med. Genet. 131A: 150-154, 2004.
NOT: KBG sendromluların 4 numaralı belirttiğim bir websiteleri mevcut. Hastalıkla ilgili bir posterleri var. Bunu Türkçeleştirmeyi ayrıca öneriyorum. İlgili kuruluşla iletişime geçip ‘’Bu posteri Türkçeleştirip kullanabilir miyiz’’ şeklinde sorulmalı. İlgili posteri aşağıya bırakıyorum. Mailde de ilgili posterin PDF halini göndereceğim, PDF çözünürlüğü yüksek.
Ürofasiyal sendrom ilk olarak 1960’larda ürolojik cerrah ve Kolombiya,
Güney Amerika’dan araştırmacı Dr. Bernardo Ochoa tarafından
tanımlanmıştır. Bozukluk Ochoa sendromu olarak da bilinir ve ilk başta
bölgeye özgü olan yerel bir bozukluk olduğuna inanılıyordu. Ürofasiyal
sendrom o zamandan beri dünya çapında ülkelerde ve etnik gruplarda
tanımlanmıştır.
Ek anormallikler, böbreklerin ve pelvisin (piyelonefrit) iltihaplanması,
idrarı böbrekten mesaneye (vezikoüreteral reflü) taşıyan tüplere geri akışını
ve kabızlığı içerebilir. Hemen tanı ve tedavi, ciddi, geri dönüşü olmayan
mesane ve böbrek hasarını azaltabilir veya potansiyel olarak
önleyebilir. Akıl etkilenmez. Ürofasiyal sendrom, hastalığın
bozulması veya değişikliklerinden (mutasyonlar) kaynaklanabilir.HPSE2 geni
veya LRIG2 geni ve otozomal resesif bir şekilde kalıtsaldır.
Üriner anormallik, doğumda ortaya çıkabilecek idrar yollarının obstrüktif bir hastalığıdır (konjenital obstrüktif üropati). Bu üropati, mesane ve omurilik arasındaki sinir sinyallerinin başarısız olması nedeniyle mesanenin eksik boşalmasına (işeme) neden olur (nörojenik veya nöropatik mesane). Mesanenin en alt noktasında, idrarın çıkarıldığı tübüler yapı olan üretraya açılan dairesel bir kas lifi bandı (üretral sfinkter) vardır. Mesane idrarla dolduğunda, normalde omuriliğe sinyaller gönderilir. Daha sonra üretral sfinkterin gevşemesine neden olan sinir sinyalleri geri gönderilir. Ve mesane büzülür ve idrar yoluyla idrar gönderir. Bununla birlikte, etkilenen bireylerde, bilinmeyen nedenlerle bu tür sinir sinyallerinin başarısız olması sözkonusudur.
Ürofasiyal sendrom, iki farklı genden, HPSE2 geni veya LRIG2 geni mutasyonlarından
kaynaklanır. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan
proteinler oluşturmak için talimatlar sağlar. Bir genin mutasyonu meydana
geldiğinde, protein ürünü hatalı, verimsiz veya olmayabilir. Belirli
proteinin işlevlerine bağlı olarak, bu vücudun birçok organ sistemini
etkileyebilir.
Ürofasiyal sendromlu bazı bireylerde, bilinen iki hastalık geninin
hiçbirinde mutasyon yoktur, bu da henüz tanımlanmamış ek genlerin de bazı
durumlarda bozukluğa neden olabileceğini düşündürmektedir.
Ürofasiyal sendrom otozomal resesif paternde kalıtsaldır. Resesif
genetik bozukluklar, bir birey, durum için anormal genin iki kopyasını, her bir
ebeveynden bir tane miras aldığında ortaya çıkar. Bir kişi hastalık için
bir normal gen ve bir gen alırsa, kişi hastalık için bir taşıyıcı olacaktır,
ancak semptom göstermeyecektir. İki taşıyıcı ebeveynin hem kusurlu geni
geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte%
25’tir. Her hamilelikte ebeveyn gibi taşıyıcı bir çocuk sahibi olma riski%
50’dir. Bir çocuğun her iki ebeveynden de normal gen alma ve bu özellik
için genetik olarak normal olma şansı% 25’tir. Risk erkekler ve kadınlar için aynıdır.
Belirti ve Semptomlar
“Ürofasiyal” teriminin belirttiği gibi, bozukluk idrar ve yüz problemleri
ile karakterizedir. Etkilenen bebeklerde ortaya çıkabilen ilk bulgu,
alışılmadık bir “tersine çevrilmiş” yüz ifadesidir. Etkilenen bebekler
gülmeye veya gülümsemeye çalıştığında, yüz kasları “ters çevirir”, böylece
ekşitmeden veya ağlıyor gibi görünürler. Ürofasiyal sendromun semptomları
ve şiddeti, aynı ailenin üyeleri arasında bile büyük ölçüde değişebilir.
Nörojenik mesane, normal olarak idrarı böbreklerden mesaneye (üreterler)
getiren tüplere idrarın geri akışına (reflü) yol açabilir; üreterlerde
(hidroüretre) ve böbreklerde (hidronefroz) anormal bir şişlik (distansiyon) ve
idrar birikmesi. Etkilenen kişilerde hidroüreter ve hidronefroz hafif ila
şiddetli arasında değişebilir. Bu tür anormalliklerin erken bir belirtisi,
uyku sırasında gündüz ve / veya gece idrar tutamamayı (günlük ve gece
enürezisi) içerebilir.
İdrar yollarının obstrüktif anormallikleri, idrar yolu hasarına ve
tekrarlayan idrar yolu enfeksiyonlarına neden olabilir. Bazı durumlarda,
bu tür enfeksiyonlar hiçbir belirtiye (asemptomatik) neden
olabilir. Bununla birlikte, diğer durumlarda, sık idrara çıkma dürtüsü, aşırı
miktarda idrarın (poliüri) geçişi, aşırı susuzluk (polidipsi), idrar geçerken
yanma hissi, ağrılı veya zor idrara çıkma (ağrılı veya zor idrara çıkma) gibi
çeşitli belirtiler ortaya çıkabilir (dizüri), mesane kontrolünün kaybı
(inkontinans), genel bir hastalık hissi (halsizlik), ateş, alt karın ve / veya
bellerde ağrı ve / veya şiddetli enfeksiyonlarda kan varlığı (hematüri) ve /
veya idrarda irin. İdrar yolu enfeksiyonu, kan dolaşımına yayılabilir, bu
da ürosepsis olarak bilinen ciddi bir komplikasyondur. Uygun tedavi
olmadan, tekrarlayan idrar yolu enfeksiyonları ve böbrek yolunda kronik
hasar nihayetinde kronik böbrek yetmezliğine yol açabilir. Böbrek
yetmezliği, böbrekler atık ürünleri idrar yoluyla atma, vücuttaki tuz ve su
dengesini düzenleme ve diğer hayati işlevlerini yerine getirme yeteneklerini
kaybettiğinde ortaya çıkar. Sonunda böbrek yetmezliği gelişen etkilenen
bireylerin tam oranı bilinmemektedir, ancak erken tedavi bu sonucun olasılığını
azaltabilir. Bozukluğu olan bazı kişilerde böbrek hasarı nedeniyle yüksek
tansiyon (hipertansiyon) olabilir. Bu ayrıca daha fazla böbrek hasarını
önlemek için aktif tedavi gerektirir. vücuttaki tuz ve su dengesini
düzenlemek ve diğer hayati işlevlerini yerine getirmek. Sonunda böbrek
yetmezliği gelişen etkilenen bireylerin tam oranı bilinmemektedir, ancak erken
tedavi bu sonucun olasılığını azaltabilir. Bozukluğu olan bazı kişilerde
böbrek hasarı nedeniyle yüksek tansiyon (hipertansiyon) olabilir. Bu
ayrıca daha fazla böbrek hasarını önlemek için aktif tedavi
gerektirir. vücuttaki tuz ve su dengesini düzenlemek ve diğer hayati
işlevlerini yerine getirmek. Sonunda böbrek yetmezliği gelişen etkilenen
bireylerin tam oranı bilinmemektedir, ancak erken tedavi bu sonucun olasılığını
azaltabilir. Bozukluğu olan bazı kişilerde böbrek hasarı nedeniyle yüksek
tansiyon (hipertansiyon) olabilir. Bu ayrıca daha fazla böbrek hasarını
önlemek için aktif tedavi gerektirir.
Etkilenen bireylerin yaklaşık üçte ikisi dışkının seyrek veya eksik
geçişi yaşayabilir (kabızlık). Bazı çocuklar bağırsak hareketlerinin
başarısız olmasının dışkının kolonda veya rektumda birikmesine neden olan bir
durum olan şifrelemeyi geliştirebilir.
Etkilenen bazı kişiler, gece lagoftalmi olarak bilinen bir durum olan uyku sırasında göz kapaklarını tamamen kapatamazlar. Bu, uyanma üzerine kuru göz hissine, hafif veya şiddetli bir duyuma neden olabilir. Lagoftalmi, kornea iltihabı (keratit), çizik kornea (kornea aşınması), enfeksiyon ve kötü uyku gibi çeşitli komplikasyonlara neden olabilir.
Ürofasiyal sendrom her iki cinsiyeti de eşit olarak etkiler. Tıbbi
literatürde 150’den fazla vaka bildirilmiştir. Literatüre göre, ailelerin
çoğunluğu Kolombiya’dan gelmektedir, ancak etkilenen aileler ABD, Birleşik
Krallık, Kuveyt, Danimarka ve İspanya’da da rapor edilmiştir.
Ürofasiyal sendrom gibi son derece nadir görülen bozukluklar genellikle
tanınmadığından, bu bozukluklar yetersiz teşhis edilir, bu da genel
popülasyondaki bu tür bozuklukların gerçek sıklığını belirlemeyi zorlaştırır.
Bazı araştırmacılar mesane, hidroüretre, hidronefroz ve ilişkili
semptomlarda anormallikler yaşayan birçok bebek ve çocuğun ürofasiyal sendrom
olabileceğinden şüphelenmektedir. Bununla birlikte, son araştırmalar,
karakteristik yüz bulguları olmayan, fonksiyonel olmayan mesanesi olan
etkilenen bireylerin, ürofasiyal sendrom kategorisinde olmayabilir, çünkü
genetik kalıpları tipik olarak farklıdır.
Kalıtım Paterni / Deseni
Ochoa sendromu da denilen Urofacial syndrome, idrar sorunları ve
olağandışı yüz ifadeleri ile karakterize bir hastalıktır.
Bu durum otozomal resesif paternde kalıtsaldır, yani her
hücredeki genin her iki kopyasının mutasyonları vardır. Otozomal resesif
koşulu olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir
kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını
göstermezler.
Ochoa sendromuyla ilişkili idrar problemleri tipik olarak erken çocukluk veya ergenlik döneminde belirginleşir. Bu bozukluğu olan insanlar, idrar akışını (inkontinans) kontrol etmekte zorluk çekebilir ve bu da yatak ıslatmasına neden olabilir. Ochoa sendromlu bireyler mesaneyi tamamen boşaltamayabilir, genellikle idrarın normalde her böbrekten mesaneye ( üreterler) taşınan kanallara geri döndüğü bir durum olan vezikoüreteral reflü ile sonuçlanabilir.). İdrar da böbreklerde birikebilir (hidronefroz). Vezikoüreteral reflü (Görsel 3) ve hidronefroz, idrar yolu ve böbrek iltihabı (piyelonefrit) sık enfeksiyonlarına yol açabilir ve sonunda böbrek yetmezliğine neden olabilecek hasara neden olabilir.
Ochoa sendromlu bazı
kişilerde HPSE2 geninde mutasyon yoktur . Bu
kişilerde, bozukluğun nedeni bilinmemektedir.
Urofacial ochoa syndrome hastalığında prevelans bilinmemektedir.
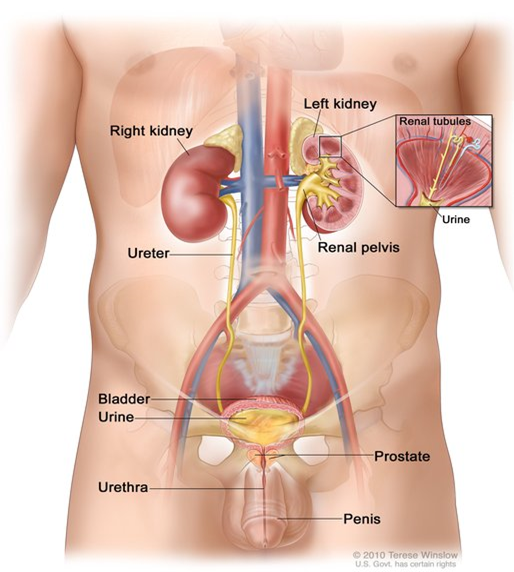
Erkek üriner sistem: Böbrekleri, üreterleri, mesaneyi ve üretrayı gösteren erkek
üriner sisteminin anatomisi. İdrar böbrek tübüllerinde yapılır ve her
böbreğin böbrek pelvisinde toplanır. İdrar böbrekten üreterler yoluyla
mesaneye akar. İdrar, vücudu üretradan terk edene kadar mesanede saklanır.
TEŞHİS YÖNTEMLERİ ve
TEDAVİLERİ
Teşhis
Ochoa sendromlu bireyler, genellikle yüz ifadesinin ters çevrilmesi
olarak tanımlanan, gülümsemeye veya gülmeye çalıştıklarında, kaşlarını çatlak
karakteristik bir yüz buruşturma sergilerler. Bu özellik idrar yolu
semptomlarından daha erken görünse de, belki bir bebek gülümsemeye başlar
başlamaz, genellikle tıbbi müdahaleye getirilmez.
Ürofasiyal sendrom için resmi tanı kriterleri
belirlenmemiştir. Tanı, karakteristik bulguların tanımlanması, ayrıntılı
bir hasta ve aile öyküsü, kapsamlı bir klinik değerlendirme ve çeşitli özel
testlere dayanarak şüphelenilir. Doktorlar, özellikle çocuk doktorları,
nefrologlar ve ürologlar, üriner işeme anormalliklerinin ve bu bozukluğu
karakterize eden benzersiz yüz ifadesinin ilişkisinin farkında olmalıdır.
Doğumda, etkilenen bebeklerde görünen ilk özellik “ters çevrilmiş” yüz
ifadesidir. Bu özelliğin güçlü bir ilişkisi ve ürolojik anormallikler
nedeniyle, ters yüz ifadelerinin varlığı idrar yolunun hızlı ve kapsamlı bir
incelemesine yol açmalıdır. Bu değerlendirme ürofasiyal sendromun erken
teşhisine ve spesifik ilişkili özelliklerin (örn. İdrar yolu enfeksiyonu) erken
teşhis edilmesine ve uygun önleyici adımların ve / veya hızlı tedavinin
sağlanmasında önemli bir rol oynayabilir.
Bu tür ürolojik değerlendirme sırasında, idrar yolu içindeki organların
yapısını incelemek ve işlevini değerlendirmek için özel görüntüleme teknikleri
kullanılabilir (örn. Böbreklerin, mesanenin, üreterlerin farklı
kısımları). Bu tür testler, özel bir kontrast ortamının enjekte edildiği
ve x-ışınlarının alındığı intravenöz piyelografiyi (IVP) ve idrar yolundan
sıvıların hareketini ve akışını inceleyen ürodinamik değerlendirmeyi
içerebilir.
Nöropatik mesaneye ve obstrüktif anormalliklere ek olarak, bu tür özel
görüntüleme çalışmaları da idrar yolunun yapısal anormalliklerini ortaya
çıkarabilir. Mesanenin anormal, ankraj bantları ve bağ dokusu kordonları
(trabekülasyon) olabilir ve mesanenin kasından küçük keseye benzer çıkıntılara
(herniler) ve aşırı kese benzeri çıkıntılara (herniler) neden olabilir. duvar
(divertikül). Ek olarak, mesanenin dış kas tabakası (detrusor kası)
alışılmadık derecede artmış refleks reaksiyonları (hiperrefleksi) ve anormal,
uzun süreli kasılmalar (hipertonik kontraktürler) gösterebilir; mesanenin
boynu anormal şekilde büyüyebilir (hipertrofik); ve / veya üretra düzensiz
spazmlara (spastik üretra) sahip olabilir ve anormal bir çapa (kalibre) sahip
olabilir.
İdrar yolu enfeksiyonları, klinik değerlendirme, mikroskobik inceleme ve
idrar örneklerinin bakteriyolojik kültürü ile teşhis edilebilir.
Moleküler genetik test, ürofasiyal sendrom tanısını
doğrulayabilir. Moleküler genetik test, hastalığa spesifik genlerde
(yani HPSE2 veya LRIG2 ) mutasyona neden olduğu
bilinen mutasyonları tespit edebilir , ancak sadece uzmanlaşmış
laboratuvarlarda bir teşhis servisi olarak mevcuttur.
Doğum öncesi tanı ürofasiyal sendrom öyküsü olan ve hastalığa neden olan
gen mutasyonunun bilindiği ailelerde mümkündür.
Tedavi
Ürofasiyal sendromun tedavisi, her bir bireyde belirgin olan spesifik
semptomlara yöneliktir. Tedavi, bir uzman ekibinin koordineli çabalarını
gerektirebilir. Çocuk doktorları, idrar yolu bozukluklarının teşhisi ve
tedavisi konusunda uzmanlaşmış doktorlar (ürologlar), böbrek bozuklukları
(nefrologlar) konusunda uzmanlaşmış doktorlar, cerrahlar, diyetisyenler ve /
veya diğer sağlık uzmanları, etkilenen bir çocuğun sistematik ve kapsamlı bir
şekilde planlanması gerekebilir tedavisi. Genetik danışmanlık etkilenen
bireyler ve aileler için faydalıdır.
Ürofasiyal sendromun hemen tanınması ve erken tedavisi, ciddi vakalarda
ortaya çıkabilecek potansiyel olarak ciddi, geri dönüşümsüz mesane ve böbrek
hasarının azaltılması veya önlenmesinde kritik öneme sahiptir.
İdrar yolu enfeksiyonlarının tedavisi genellikle bakteriyel
enfeksiyonların ve ağrı kesicilerin (örn. Analjezikler) tedavisi ve önlenmesi
için antibiyotik içerir. Bazı insanlarda, idrar yolu tıkanıklığını ortadan
kaldırmak ve idrar yolunun belirli kısımlarını (örn. Üreterler,
üreterovesiküler kavşak) yeniden yapılandırmak için ameliyat
yapılabilir. Yapılan cerrahi prosedürler anatomik anormalliklerin
şiddetine ve ilişkili semptomlarına bağlıdır.
Ürofasiyal sendromlu bireyleri tedavi etmek için spesifik ilaçlar,
anti-kolinerjik ve alfa-1-adrenerjik blokerler kullanılmıştır. Kabızlık,
genel popülasyonda olduğu gibi standart kılavuzlarla tedavi edilmelidir.
Mesanenin tam boşaltımı sağlanamadığı durumlarda, Aralıklı
kateterizasyon (T.A.K. temiz aralıklı kateterizasyon) gerekebilir. Üst üriner
sistem bozulmasını ve böbrek yetmezliğini önlemek için erken tanı ve tedavi
gereklidir.
Kronik böbrek yetmezliği yaşayan etkilenen çocuklarda, tedavi
seçenekleri, fazla atık ürünleri düzenli olarak kandan (diyaliz) uzaklaştıran
belirli prosedürleri içerebilir. Bu prosedürler, bir makine yoluyla kanı
süzerek (hemodiyaliz) ve / veya vücudun karnında doğal bir filtreleme membranı
(periton diyalizi) kullanarak atıkların giderilmesini içerebilir. Bazı
ciddi böbrek yetmezliği vakalarında böbrek nakli düşünülebilir.
Etkilenen çocukların potansiyellerine ulaşmasını sağlamak için erken
müdahale önemlidir. Yararlı olabilecek özel hizmetler, özel sosyal desteği
ve diğer tıbbi ve / veya sosyal hizmetleri içerebilir.
Hastalıkla İlişkili Genler
Aşağıdaki bozuklukların belirtileri ürofasiyal sendromdakilere benzer
olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Hinman-Allen sendromu veya nörojenik olmayan nörojenik mesane olarak da
bilinen Hinman sendromu, nörolojik bir eksiklik olmadığı için nöropsikolojik
kökenli olduğuna inanılan nadir bir işeme bozukluğudur. Etkilenen
bireyler, ortaya çıkmayan yüz ifadesinde anormallikler dışında, ürofasiyal
sendromlu bireylerde görülenlere oldukça benzer klinik özellikler sergiler.
Ürofasiyal sendroma ek olarak, ek konjenital bozukluklar nörojenik
mesane, hidroüretre ve / veya hidronefroz gibi idrar yolu
anormallikleri; etkilenen erkeklerde kriptorşidizm gibi genital sistem
malformasyonları; yüz anormallikleri; ve / veya ürofasiyal sendromla
potansiyel olarak ilişkili olanlara benzer diğer fiziksel anormallikler.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}