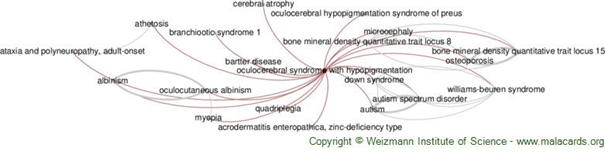
Hipopigmentasyonlu Oküloserebral Sendromu, derinin ve saçın normal renginin (hipopigmentasyon) ve beynin gözlerini ve bazı kısımlarını etkileyen merkezi sinir sisteminin anormallikleri (oküloserebral) ile karakterize ve oldukça nadir görülen kalıtsal bir hastalıktır.
Belirti ve Semptomlar
Cross Sendromu olarak da bilinen
Hipopigmentasyonlu Oculocerebral Sendrom, doğuştan veya erken bebeklik
döneminde ortaya çıkabilen oldukça nadir görülen kalıtsal bir hastalıktır.
Hastalığın ilk gözle görülür belirtileri, hipopigmentasyon veya cilt ve saçın
toplam renk eksikliğidir (depigmentasyon). Cilt güneşe ve ışığa maruz kalmaya
karşı aşırı hassas olabilir. Çoğu durumda, saç doğumda genellikle gümüş ya da
gümüş grisi olur. Bebeklik döneminden sonra daha ciddi semptomlar görülebilir.
Hipopigmentasyonlu Oculocerebral
sendromlu bir bebekte üç aylıkken merkezi sinir sisteminde anormallikler olabilir.bu anormallikler ; Çeşitli
kaslardaki özellikle ellerde irade dışı,anlamsız ve yavaş hareketler görülmesi,
bir veya iki gözün anormal küçüklüğü de dahil olmak üzere gözlerde
anormallikler (mikroftalmi), bazı durumlarda ışığın geçtiği gözlerin öndeki açık kısmı (kornealar) alışılmadık
derecede küçük olması (mikrokornea), göz merceklerinde (katarakt) şeffaflık kaybı (opaklık), istemli
hareketlerin koordinasyonunda zorluk çekme,baş hareketlerinde zorlanma ve
hiperekstansiyon olarak ortaya çıkabilmektedir.
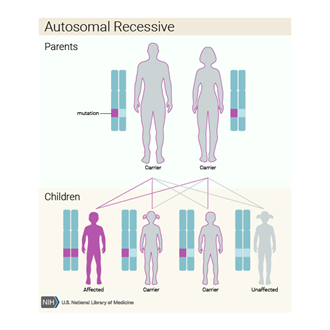
Kalıtım Paterni/Deseni,
Hipopigmentasyonlu
Oculocerebral Sendromun otozomal resesif genetik özellik olarak kalıtımsal
olduğuna inanılmaktadır.
Teşhis Yöntemleri ve Tedaviler
Hipopigmentasyonlu
Oculocerebral Sendrom’a neden olan mutant genin varlığını saptamak için
moleküler genetik test uygulanması, hastanın öyküsünün incelenmesi ve dikkatli
fizik muayenesi yapılması,hastalıkla özdeşleşmiş belirti ve semptomlara
bakılması, Hipopigmentasyonlu Oculocerebral Sendrom tanısı koymak için
izlenilen yollardır.
Hipopigmentasyonlu
Oculocerebral Sendrom Tedavileri, gözlemlenen spesifik belirti ve semptomlara
yöneliktir. Genellikle çocuk doktorları, nörologlar, dermatologlar, göz
doktorları ve diş hekimleri gibi çeşitli uzmanlıkların koordine çabalarını
gerektirir.
Etkilenen bireyler
yüksek hassasiyetli cilde sahip olabileceğinden yüksek SPF güneş kremi,
şapkalar ve uzun kollu gömlekler kullanılması önerilir, göz kusurlarını düzeltmek
için gözlükler veya kontak lensler gerekebilir.
Genetik Görülme Sıklığı
Hipopigmentasyonlu
Oculocerebral Sendrom, erkekleri ve kadınları eşit sayıda etkileyen son derece
nadir bir hastalıktır. Tıp literatüründe 15’ten az vaka bildirilmiştir. Bu
gözlenen vakaların çoğu aileler içinde meydana gelmiştir.
De
Sanctis-Cacchione sendromu, nörolojik anormallikler, zihinsel gerilik,
alışılmadık kısa boy (cücelik) ve testislerin veya yumurtalıkların az
gelişmişliği ile bağlantılı olarak ortaya çıkan xeroderma pigmentosum’un (XP)
cilt ve göz semptomları ile karakterize oldukça nadir görülen bir hastalıktır.
. Xeroderma pigmentosum, ultraviyole ışığına (ışığa duyarlılık), cilt
rengindeki renk bozukluklarına ve çeşitli göz bozuklukları ve cilt
kanserlerinin olası gelişimine karşı yüksek reaksiyon gösteren, nadir görülen
kalıtsal cilt hastalıkları grubudur. De Sanctis-Cacchione sendromu ile
ilişkili en yaygın nörolojik anormallikler düşük zeka, anormal derecede küçük
bir kafa (mikrosefali), gönüllü hareketi koordine etme kabiliyetinin kaybı
(ataksi) ve / veya yok (refleksi) veya zayıflamış (hiporefleksi)
refleksleridir.
Orpha.net
web sitesinde “Bu hastalık Xeroderma pigmentosum’a taşındı”
bilgisi karşımıza çıkmaktadır. De Sanctis-Cacchione sendromu, başlangıçta ağır
nörolojik anormallikleri olan XP vakalarına atfedilen ancak genel kullanımda
olmayan bir terimdir.
De
Sanctis-Cacchione sendromunun belirtileri, vücudun genlerin yapı taşlarına
(DNA) zarar verememesi nedeniyle ortaya çıkar. Hasar, güneş ışınları gibi
ultraviyole ışığa maruz kalmasından kaynaklanır. Herkesin bu hasarı
karmaşık bir işlemle onarabilen bazı bağ dokusu hücreleri (fibroblastlar)
vardır. Bununla birlikte, De Sanctis-Cacchione sendromundan etkilenen insanlarda
bulunan fibroblastlar, DNA’larını onarma yeteneğinden yoksundur veya
kapasiteleri azalır. Ek olarak, etkilenen bazı kişilerin hücreleri güneş
ışığına zarar veren cildi düzgün şekilde onaramaz.
Vücudun
güneş ışığına zarar veren DNA’yı onarma kapasitesine bağlı olarak çeşitli XP
formları (alt bölümler) tanımlanmıştır. XP’nin alt bölümlerinden herhangi
biri De Sanctis-Cacchione sendromunda ortaya çıkabilir. Bununla birlikte
klasik XP formu (xeroderma pigmentosum, Stype A) veya xeroderma pigmentosum, D
tipi en sık De Sanctis-Cacchione sendromu ile birlikte bulunur.
Araştırmacılar,
bazı De Sanctis-Cacchione sendromu vakalarının, kromozom 10’un (10q11) uzun
kolunda (q) bulunan belirli bir genin bozulmasından veya değişmesinden
kaynaklanabileceğini belirlemiştir. İnsan hücrelerinin çekirdeğinde
bulunan kromozomlar her bireyin genetik bilgisini taşır. İnsan
kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve erkeklerde
bir X ve bir Y kromozomu ve kadınlarda iki X kromozomu içeren ek bir 23. cinsiyet
kromozomu çiftidir. Her kromozomun “p” işaretli kısa bir kolu ve “q”
işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok
gruba bölünmüştür. Örneğin, “kromozom 10q11”, kromozom 10’un uzun
kolundaki bant 11 ile ilgilidir. Numaralı bantlar, her bir kromozomda bulunan
binlerce genin konumunu belirtir.
Belirti ve Semptomlar
De
Sanctis-Cacchione sendromunda en erken semptomlar, ultraviyole ışığına maruz
kaldıktan sonra meydana gelen aşırı çil ve kabarma dahil xeroderma pigmentosum
(XP) ile ilişkili cilt anormallikleridir (ışığa duyarlılık). Bazı
durumlarda, güneş ışığı ile temas ettikten hemen sonra ağrı ve kabarma meydana
gelebilir. Akut güneş yanığı ve cildin kalıcı kızarıklığı veya iltihabı
(eritem) de De Sanctis-Cacchione sendromunun erken belirtileridir. Çoğu XP
vakasında, bu semptomlar doğumdan hemen sonra veya üç yaşından sonra görülebilir. Bununla
birlikte, bazı nadir durumlarda, daha sonra çocukluk döneminde semptomlar
belirgin olmayabilir. De Sanctis-Cacchione sendromunun çoğu vakasında,
başlangıç genellikle bebeklik döneminde görülür.
Sıklıkla
De Sanctis-Cacchione sendromu ile ilişkili ek cilt semptomları, cildin
alışılmadık derecede koyu (hiperpigmentasyon) veya açık (hipopigmentasyon)
alanlarını içerir. Bazı durumlarda, cilt renginin tamamen kaybolması
(depigmentasyon) ve / veya aşırı skar oluşabilir. Siğil benzeri lezyonlar
(aktinik keratozlar) yanı sıra, cildin yüzeyine yakın küçük kan damarlarının
anormal şekilde genişlemesinden kaynaklanan küçük kırmızı cilt lezyonları
(telangiektaziler) gelişebilir. Cilt ayrıca zayıflayabilir ve kolayca
zarar görebilir. Dejeneratif (atrofik) değişiklikler meydana gelebilir ve
cilt kuru ve pürüzsüz görünebilir.
De
Sanctis-Cacchione sendromu olan çoğu çocuğun genellikle bir veya daha fazla
nörolojik anomalisi vardır; en sık görülen zeka düşüktür. Diğer
anormallikler, alışılmadık derecede küçük bir baş (mikrosefali); işitme
bozukluğu (duyusal sağırlık); eksik (arefleksi) veya zayıflamış
(hiporefleksi) refleksleri; ve / veya bazı kaslarda sertlik ve hareket
kısıtlılığına (spastisite) neden olan sertliğin artması. Etkilenen
bireyler ayrıca gönüllü hareketleri (ataksi) ve / veya vücudun anormal istemsiz
hareketlerini, kontrolsüz sarsıntılı hareketler (anormal yavaş hareketler
(koreotetoz)) ile birlikte koordine etme kabiliyetlerini de gösterebilir.
De
Sanctis-Cacchione sendromu, zaman zaman, serebellum (serebellar atropi) olarak
bilinen beynin bir kısmının yavaş ilerleyici dejenerasyonu ve / veya beynin
diğer kısımlarının (yani, korteks, baz pontisi) degrasyonunu içeren bir grup
ilerleyici bozukluktan herhangi biriyle ilişkilidir. ve aşağı olivary çekirdekleri. Bu
klinik tablo, kalıtsal olivopontocerebellar atrofilerde görülene
benzer. Belirtiler, bozuk kas koordinasyonu (ataksi), titreme, istemsiz
hareketler ve konuşma bozukluklarını (dizartri) içerebilir. (Bu hastalık
hakkında daha fazla bilgi için, Nadir Hastalık Veri Tabanında arama teriminiz
olarak “Kalıtsal Olivopontocerebellar Atrofi” yi seçin.)
De
Sanctis-Cacchione sendromu olan bireyler ayrıca alışılmadık derecede yavaş
gelişme, kısa boylanma (cücelik), zihinsel gerilik ve / veya testis veya
yumurtalıkların (hipogonadizm) yetersiz fonksiyonu ile sonuçlanan derin büyüme
gecikmeleri sergileyecektir.
Benign
cilt tümörleri, De Sanctis-Cacchione sendromu ile ilişkili olabilir ve beş
yaşından önce başlaması mümkündür. Bunlar, kan damarlarından (anjiyomlar) oluşan
tümörler ve / veya sıklıkla cildin güneşe maruz kalan bölgelerinde ortaya çıkan
hızla büyüyen tümörler gibi, önceden malign veya iyi huylu (kanserli olmayan)
tümörleri içerebilir.
De
Sanctis-Cacchione sendromu olan kişiler erken yaşta cilt kanseri yaşarlar. Örneğin,
malign melanom, bazal hücreli karsinom ve skuamöz hücreli karsinom gibi cilt
kanserleri bu rahatsızlığı olan kişilerde sıklıkla görülür; En sık
etkilenen bölgeler baş, boyun ve yüzdür. (Bu bozukluklar hakkında daha
fazla bilgi için bu raporun İlgili Bozuklukları bölümüne bakın.)
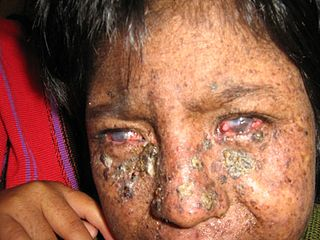
De
Sanctis-Cacchione sendromunda, XP ile ilişkili göz semptomlarından bazıları
mevcut olabilir. Bunlar ışığa aşırı derecede toleranssızlık içerebilir
(fotofobi); gözlerin kornealarının iltihabı (keratit); gözlerin beyaz
kısmını kaplayan zarı iltihabı (konjonktivit); göz kapaklarının dışa dönük
(ektropion); ve / veya göz kapaklarının içe dönük (entropion). Cilt
ve gözlerle ilgili semptomların şiddeti, ultraviyole ışığına maruz kalma
miktarına bağlı olabilir.
Genetik Görğlme Sıklığı
De
Sanctis-Cacchione sendromu, otozomal resesif bir özellik olarak
kalıtsaldır. Klasik genetik hastalıklar da dahil olmak üzere insan
özellikleri, biri babadan diğeri de anneden alınan iki genin etkileşiminin
ürünüdür.
Resesif
genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı
anormal geni aldığında ortaya çıkar. Bir birey hastalık için bir normal
gen ve bir gen alırsa, kişi hastalık için taşıyıcı olacaktır, ancak genellikle
semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de
etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Ebeveynler
gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte% 50’dir. Bir
çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak
normal olma şansı% 25’tir.
Belgelenen
vakaların yaklaşık yüzde 30’unda, De Sanctis-Cacchione sendromundan etkilenen
bireylerin, kanla ilişkili (akraba) anne-babaları olmuştur. Tüm bireyler
4-5 anormal gen taşır. Yakın akraba olan ebeveynlerin (akraba), ilişkisiz
ebeveynlerden her ikisinin de aynı anormal geni taşıması olasılığı daha
yüksektir, bu da resesif genetik bozukluğu olan çocukların görülme riskini
artırır.
ABD
ve Avrupa’da tahmini yaygınlığı 1 / 1.000.000 olup, diğer ülkelerdeki (örneğin
Japonya, Kuzey Afrika ve Pakistan), özellikle de yüksek derecede akrabalık
dereceli topluluklarda daha yüksek rakamlara sahiptir.
Kalıtım Paterni Deseni
De
Sanctis-Cacchione sendromu erkekleri ve kadınları eşit sayıda etkileyen çok
nadir görülen bir hastalıktır. Batı tıp literatüründe yaklaşık 200 vaka
bildirilmiş olmasına rağmen, bu hastalığın kesin vakalarının sayısı
bilinmemektedir. Semptomların başlangıcı genellikle yaşamın ilk yılında
ortaya çıkar, ancak nadir durumlarda erken veya geç çocukluk döneminde ortaya çıkabilir. Bazı
nörolojik semptomların başlangıcı beş ila 10 yaşları arasında veya hatta
yaşamın ikinci on yılında ortaya çıkabilir. De Sanctis-Cacchione sendromu
ilk olarak 1932 yılında tıp literatüründe tanımlanmıştır.
XP,
DNA onarımında rol alan 8 gendeki mutasyonlardan kaynaklanır. Bu genlerin
yedi, DA için XPG ( ERCC5 ), nükleotid
onarım (EŞ) yer alırlar. XPV veya POLH , UV kaynaklı
hasarı içeren DNA’yı kopyalamak için gereken DNA polimeraz etasını kodlar.
Teşhis Yöntemleri ve Tedavileri
Teşhis
De
Sanctis-Cacchione sendromunun teşhisi, xeroderma pigmentosum’un bir veya daha
fazla nörolojik anormallik, zihinsel gerilik, cücelik ve testislerin veya
yumurtalıkların yetersiz fonksiyonu ile birlikte ortaya çıktığı ortaya
çıktığında doğrulanabilir.
Doğumdan
önce (doğum öncesi) XP tanısı, amniyosentez adı verilen özel bir prosedür
kullanılarak doğrulanabilir. Bu prosedür sırasında, fetüsü çevreleyen bir
sıvı örneği alınır ve fetüsün XP olup olmadığını belirlemek için testler
yapılır. Bu prosedür genellikle XP öyküsü olan aileler için bir tarama
sürecinin bir parçası olarak yapılır.
De
Sanctis-Cacchione sendromunun diğer semptomlarının mevcut olup olmadığını
belirlemek için kapsamlı bir klinik değerlendirme yapılmalıdır (yani nörolojik
anormallikler, zihinsel gerilik, cücelik ve hipogonadizm. Bu değerlendirme,
manyetik rezonans görüntüleme (MRI) gibi nörogörüntüleme çalışmalarını
içerebilir. ve bilgisayarlı tomografi (BT) taramaları.
De
Sanctis-Cacchione sendromu tanısını doğrulamak için özel laboratuvar testleri
kullanılabilir. Bu testler, beyaz kan hücrelerinde (lenfositler),
karaciğer hücrelerinde, kornea hücrelerinde ve De Sanctis-Cacchione
sendromundan etkilenen insanlardan alınan cilt hücrelerinde hatalı DNA
onarımını tespit edebilir. Bu tür testler sırasında, hücreler UV
radyasyonuna ve / veya bazı kanser üreten maddelere (kanserojenler) maruz
kalırlar. Bu maddelere maruz kaldıktan sonra, bu hücrelerin kusurlu DNA
onarım işlemi belirginleşir.
Tedavi
De
Sanctis-Cacchione sendromu olan bireylerde, cilt lezyonlarının ve diğer
komplikasyonların (örneğin, cilt kanserleri ve bazı nörolojik semptomlar)
gelişimini önlemek için cildin güneş ışığından (örn. Topikal güneşten
koruyucular, güneş gözlükleri, çift kat giysi) tamamen korunması gerekir.
. De Sanctis-Cacchione Sendromu olan etkilenen bireyler, ultraviyole ışığa
maruz kalmamak için gündüz saatlerinde dış mekan etkinliklerini
sınırlandırmalıdır. Sigara dumanındakiler gibi kimyasal kanserojenlerden
kaçınılması da önerilmektedir.
Deri
kanseri olan kişiler için, lezyonların erken tespiti ve cerrahi olarak
çıkarılması esastır. Derinin ve gözlerin uzmanlar tarafından düzenli
muayenesi önerilir. Genetik danışmanlık, etkilenen bireyler ve aileleri
için faydalı olacaktır. Diğer tedavi semptomatik ve destekleyicidir.
XP için bir tedavi yoktur, ancak herhangi bir cilt kanserini
değerlendirmek ve tedavi etmek için güneşten korunma ve düzenli takip yaşam
süresini uzatır. Nörolojik hastalığı ve titiz UV korumasına sahip
olmayanlar için prognoz iyidir. Bununla birlikte, nörolojik anormallikler
ilericidir ve ömrünün kısalmasına neden olabilir.
Araştırma Terapileri
De
Sanctis-Cacchione sendromunu tedavi etmek için kozmetik cerrahi prosedür
(dermabrazyon), kimyasal peeling, tümör eksizyonu ve / veya yüz cilt grefti
kullanılmıştır. Dermabrazyon ve topikal 5-flüoroürasil, premalign veya
erken cilt lezyonlarının tedavisinde etkili olabilir. Bir katalaz kreminin
uygulanması bazı çocuklarda tümörleri önlüyor gibi görünmektedir. A
vitamini türevlerini içeren merhemler veya kremler de incelenmektedir.
Kapsüllenmiş
T4 endonükleaz VB lipozomu (T4N5), cilt kanserlerini ve xeroderma pigmentosum
ile ilişkili diğer cilt anormalliklerini önlemek için kullanılan bir yetim
ilaçtır. İlk çalışmalar, topikal T4N5 ile tedavinin, bazen De Sanctis-Cacchione
sendromu ile bağlantılı cilt kanseri gelişim hızını yavaşlattığını
göstermiştir. Xeroderma pigmentosum tedavisinde bu ilacın uzun vadeli
güvenliğini ve etkinliğini belirlemek için daha fazla çalışmaya ihtiyaç
vardır.
De Sanctis-Cacchione sendromu ile ilişkili göz
anormalliklerinin tedavisi için çeşitli tedaviler
incelenmektedir. Ameliyat, gözleri etkileyen problemler nedeniyle sıklıkla
mümkün olmamakla birlikte, korneanın bir kısmının çıkarıldığı ve değiştirildiği
(keratoplasti) özenle seçilmiş kişilerde çalışmalar yürütülmektedir. Yapay
gözyaşı ve yumuşak kontakt lensler de kullanılmıştır.
Hastalıkla İlişkili Genler
Aşağıdaki
bozuklukların belirtileri De Sanctis-Cacchione sendromununkilerle benzer
olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Cockayne
sendromu, kısa boy ile karakterize nadir görülen kalıtsal bir
hastalıktır; ayırt edici cilt anormallikleri; karakteristik
kraniyofasiyal malformasyonlar ve erken yaşlanmış (progerik) bir
görünüm; görme bozukluğu ile sonuçlanan göz
anormallikleri; sağırlık; ve / veya zihinsel geriliği. Bozukluğu
olan bireylerde, göz anormallikleri, göz sinirlerinin kademeli olarak
bozulmasını (optik atrofi), gözleri kaplayan sinir bakımından zengin zarın
ilerleyen dejenerasyonunu (retina dejenerasyonu) ve / veya göz merceklerinin
anormal şekilde bulanıklaşmasını içerebilir. (katarakt). Ek olarak, cilt
güneş ışığına karşı anormal derecede hassas olabilir; bu da yara izi, pullu
döküntü, anormal pigmentasyon ve etkilenen alanların dejenerasyonu (atrofi) ile
sonuçlanabilir. Cockayne sendromu olan bireyler ayrıca erken burun
görünümüne ve ince burun, batık gözler ve / veya çıkıntılı alt çene
(prognatizm) dahil olmak üzere karakteristik yüz anormalliklerine sahip
olabilir. Etkilenen bireyler, aynı zamanda nadir görülen saçı ve çeşitli
iskelet malformasyonlarını içeren ek fiziksel anormalliklere de sahip
olabilir. Cockayne sendromu, otozomal resesif bir özellik olarak
kalıtsaldır. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık
Veritabanında arama teriminiz olarak “Cockayne” ı seçin.)
Pigmentli
kserodermoid, kseroderma pigmentozuma benzer semptomların geç başlangıcı ile
karakterize bir durumdur. Bu belirtiler güneş ışığına yüksek reaksiyon
gösterebilir (ışığa duyarlılık); cilt renk değişikliği; küçük kırmızı
cilt lezyonları (telangiektazi); göz anormallikleri; ve / veya iyi
huylu (kanserli olmayan) tümörler (anjiyomlar ve
keratoakanttomlar). Etkilenen bireyler bazı cilt kanseri türlerinin erken
yaşlarında karşılaşabilirler (örneğin, bazal hücreli karsinom, skuamöz hücreli
karsinom, malign melanom). Pigmentli xerodermoid olan kişiler ultraviyole
ışıktan zarar gören DNA’yı onarabilir; Ancak, onarım sonrası işlem
hatalı. Pigmentli kserodermoidin nedeni bilinmemektedir. Bununla
birlikte, bazı bilim adamları, otozomal resesif bir özellik olarak kalıtsal
olabileceğine inanmaktadır.
Aşağıdaki
bozukluklar, ikincil özellikler olarak De Sanctis-Cacchione sendromu ile
ilişkilendirilebilir. Ayırıcı tanı için gerekli değildir:
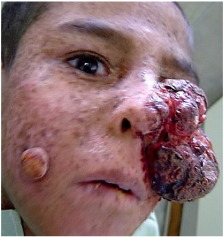
Malign
melanom, cildin üst tabakasının melanin hücrelerinden (epidermis) veya mollerde
(nevi) bulunabilen benzer hücrelerden gelişen bir cilt kanseri
türüdür. Erken evrelerde, çoğu melanom herhangi bir spesifik semptom
üretmez. Daha sonra iyileşmeyen lezyonlar veya boyut veya renkte
değişiklikler gösteren mevcut moller olarak görünebilirler. Bu tip cilt
kanseri nihayetinde düşük cilt seviyelerine ve bitişik dokulara yayılır ve
vücudun hayati organlarında yeni tümör büyümelerine neden olabilir. (Bu
hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama
teriminiz olarak “Melanom” seçeneğini seçin.)
Bazal
hücreli karsinomlar cilt yüzeyinde ortaya çıkan tümörlerdir. Küçük,
parlak, sert doku kütleleri (nodüller); yassı, yara benzeri lezyonlar
(plaklar); veya kalın, kuru, gümüş pullarla kaplı kırmızı
lekeler. Eğer tedavi edilmezse, bazal hücreli karsinom vücudun diğer
bölgelerine yayılabilir (metastaz yapmaz). Bu tip cilt kanseri, biyopsi
olmadan sedef hastalığından veya lokalize dermatitten ayırt edilmesi zor
olabilir. Güneş ışığına veya iyonlaştırıcı radyasyona aşırı maruz kalmak
bu tür cilt kanserine neden olabilir.
Skuamöz
hücreli karsinomlar, genellikle cildin güneşe maruz kalan bölgelerinde ortaya
çıkan, ancak vücudun herhangi bir yerinde görülebilen tümörlerdir. Bu cilt
kanseri biçimi, pullu veya kabuklu bir yüzeye sahip görünen lezyonlarla
karakterizedir. Bu lezyonlar altta yatan dokulara
yayılabilir; Bununla birlikte, uygun tedavi ile, Squamous hücreli karsinom
genellikle tedavi edilebilir. (Bu hastalık hakkında daha fazla bilgi için,
Nadir Hastalık Veritabanında arama teriminiz olarak “Squamous Cell Carcinoma”
seçeneğini seçin.
Pachyonychia congenita , öncelikle cildi ve tırnakları etkileyen bir durumdur.
Bu durumun belirti ve semptomları genellikle yaşamın ilk birkaç yılında
belirginleşir.
Pachyonychia konjenita ile hemen hemen herkes , tırnak
ve ayak tırnaklarının kalınlaşmasına ve anormal şekilde şekillenmesine neden
olan bazı hipertrofik tırnak distrofisi belirtileri gösterir. Etkilenen
çocukların çoğu, ayak tabanlarında ve daha az yaygın olarak ellerin avuçlarında
çok ağrılı kabarcıklar ve nasırlar geliştirir. Bu durum palmoplantar
keratoderma olarak bilinir. Ayaklardaki şiddetli kabarcıklar ve nasırlar
genellikle ilk çocukluk döneminde yürümeye başladıklarında oluşmaya başlar ve
yürümeyi acı verici veya imkansız hale getirebilir.
Pachyonychia congenita , etkilenen bireyler arasında
çeşitlilik gösteren çeşitli ek özelliklere sahip olabilir. Bu özellikler
arasında dil üzerinde ve yanakların içindeki (beyaz lökokeratoz) kalın beyaz
lekeler; dirsek, diz ve bel üzerinde saç köklerinin çevresinde gelişen
foliküler keratozlar; koltuk altı, kasık, sırt veya kafa derisindeki kistler;
ve avuç içi ve tabanlarda aşırı terleme (palmoplantar hiperhidrozu). Etkilenen
bazı kişiler ayrıca, normalde cildi ve saçı yağlayan sebum denilen yağlı bir
madde ile doldurulmuş steatositoma adı verilen yaygın kistler de geliştirir.
Pakyonychia congenitalı bazı bebeklerde doğum öncesi veya doğum sonrası dişler
vardır. Görsel1 doğumda veya erken bebeklik döneminde var olan
dişlerdir. Bazı durumlarda pachyonychia congenita ses kutusunu etkileyebilir (
larinks) ses kısıklığına veya solunum problemlerine neden olur.
Araştırmacılar, pachyonychia congenita’yı PC-1 veya
PC-2 olmak üzere iki tipten biri olarak ayırt etmek için kullanıldı , genetik
neden ve belirti ve semptomların paternine dayanarak. Bununla birlikte, daha
fazla etkilenen birey tanımlandıkça, iki türün özelliklerinin önemli ölçüde üst
üste geldiği açıkça ortaya çıktı. Şimdi araştırmacılar pachyonychia
congenita’nın değiştirilen geni temel alan bir tanımını tercih ediyorlar .
Not: EBS,
PC’li küçük çocuklarda yanlış tanı alabilir, çünkü kabarcık oluşumuna karşı
daha fazla eğilimli ve keratodermaya karşı daha az eğilimlidirler.
Genetik Değişiklikler /Etken Faktörler
KRT6A , KRT6B , KRT6C , KRT16 ve KRT17 dahil olmak üzere birçok gendeki mutasyonlar pachyonychia
congenita’ya neden olabilir . Bu genlerin tümü keratinler adı verilen sert,
lifli proteinleri yapmak için talimatlar sağlar.. Bu proteinler, cildi, saçı ve
tırnakları oluşturan dokulara güç ve esneklik sağlayan ağlar oluşturur.
Pakyonychia konjenita KRT6A genindeki mutasyonlardan
kaynaklandığı zaman PC-K6a olarak sınıflandırılır. Benzer şekilde, KRT6B gen
mutasyonları PC-K6b’ye, KRT6C gen mutasyonları PC-K6c’ye, KRT16 gen
mutasyonları PC- K16’ya ve KRT17 gen mutasyonları PC- K17’ye neden olur.
Keratin genlerindeki mutasyonlar, bu proteinlerin
hücreler içinde güçlü, kararlı ağlar oluşturmasını önleyen keratin
proteinlerinin yapısını değiştirir. Bu ağ olmadan cilt hücreleri kırılgan hale
gelir ve kolayca zarar görebilir, bu da cildi sürtünmeye ve küçük travmaya
karşı daha az dirençli hale getirir. Yürüme gibi normal aktiviteler bile cilt
hücrelerinin parçalanmasına neden olabilir ve bu da ciddi, ağrılı kabarcıklar
ve nasırlar oluşmasına neden olabilir. Arızalı keratinler ayrıca saç
foliküllerinde ve tırnaklarındaki hücrelerin büyümesini ve fonksiyonunu da
bozar ve pachyonychia congenita’nın diğer özelliklerine neden olur .
Belirti ve Semptomlar
PC’nin baskın klinik özelliği olan hipertrofik tırnak distrofisi, tipik olarak yaşamın ilk birkaç ayında ve birkaç yıl içinde kaydedilmesine rağmen, nadir durumlarda yaşamda daha sonra ortaya çıkar. Tırnak distrofisinin iki fenotipe düşmesi gibi görünüyor:
– Tam uzunlukta yetişen ve belirgin distal
hiperkeratozun neden olduğu yukarı meyilli olan tırnaklar (genellikle çivinin
vurgulanmış eğriliği ile)
– Hiperkeratozun hafifçe eğimli bir distal bölgesini ve
açıkta kalan distal parmak ucunu bırakarak erken sonlanan bir tırnak plakasına
sahip tırnaklar
Fokal palmoplantar keratoderma, genellikle bir çocuğun ağırlık taşımaya ve yürümeye başladığında yaşamın ilk birkaç yılında ortaya çıkar. Kabarcıklar yoğun ağrı ile sonuçlanan keratodermanın altında gelişir. Birçok insan için, kabarcıklar ve sürekli ayak ağrısı, sıcak havalarda soğuk havalardan daha şiddetlidir. Plantar fokal kabarma ile bağlantılı ağrı, koltuk değneği, bastonlar veya tekerlekli sandalyelerin kullanılmasını gerektirebilir. Nadiren, keratoz palmoplantaris transgrediens (hiperkeratozun palmar ve / veya plantar cildin ötesinde bitişik uzantısı) mevcuttur.
– Avuçlarında oluşabilecek değişken şiddette keratoderma
olarak tanımlanan fokal epidermolitik olmayan palmoplantar keratoderma
(FNEPPK), daha önce belirgin bir varlık olarak kabul edildi, ancak şimdi
spektrumun bir parçası olarak kabul edildi. PC
Oral lökokeratoz(dil ve yanakta kalınlaşmış beyaz lekeler) sıklıkla bulunur. Bebeklerde oral lökokeratoz Candida albicans olarak yanlış tanı alabilir ve emilmesinde zorluklara neden olabilir.
Bazı kişilerde genellikle dirseklerde, dizlerde veya gövdede folikülerkeratoz görülür. Geç çocukluk ve gençlik yıllarında daha sık görülür ve yetişkinlerde daha az sorunlu hale gelir.
Yaygın steatositomalar / steatosistleri (iyi huylu lezyonlar) ve vellus kıllarını içeren pilosebasöz kistler. Ergenlikte kistler sayıca artabilir. Erken başlangıçlı bildirilmiştir [ Feng et al 2003 ] ve Uluslararası Pachyonychia Congenita Araştırma Kayıt Defteri’ne (IPCRR) kaydedilir.
– İnce tırnak tutulumu ile ergenlikte gelişebilecek,
ancak palmoplantar keratoderma içermeyen yaygın steatositomalar olarak
tanımlanan Steatositoma multipleks (SM), KRT17’de heterozigos patojenik
varyantları ile birlikte ortaya çıkmaktadır
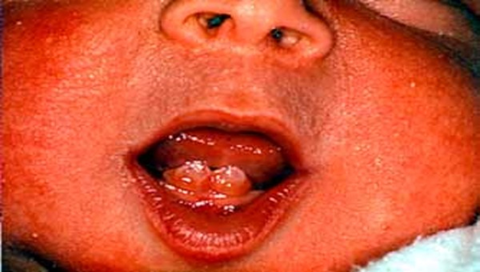
Doğumsal dişler veya doğum öncesi dişler. Bazı bireylerde birkaç doğum öncesi ya da doğumsal diş olmasına rağmen, bu bulgu aynı aile içinde bile tutarlı bir şekilde mevcut değildir [ Leachman et al 2005 ]. Doğumsal dişler genellikle KRT17’de patojenik varyantlarla ilişkilidir . Birincil ve ikincil dişçilik normaldir.
Oluşabilecek Diğer Bulgular :
– Bireylerin yaklaşık% 50’sinde gözlenen avuç içi ve
tabanların aşırı terlemesi (palmoplantar hiperhidroz)
– Aksiller ve inguinal kist oluşumu
– Kulakta aşırı miktarda mumsu üretimi
– Şiddetli ve açıklanamayan kulak ağrısı
– Ses kısıklığı (laringeal tutulum), öncelikle küçük
çocuklarda bildirilmiştir. Nadir olmasına rağmen, laringeal tutulum müdahale
gerektiren hayatı tehdit edici solunum sıkıntısına neden olabilir.
– Bazen sekonder olarak enfekte olan açısal cheilitis
(ağız açısında yanma ve yanma)
– Tırnakların altında belirgin ödem (ve ara sıra
kabarcık oluşumu) olan Paronişi; lenfatik uzama gösterebilir ve bazen
enfeksiyondan kaynaklanabilir
Pakyonychia konjenita prevalansı bilinmemekle
birlikte, dünya çapında
etkilenebilecek binlerce insanın nadir olarak görüldüğü bir hastalıktır.
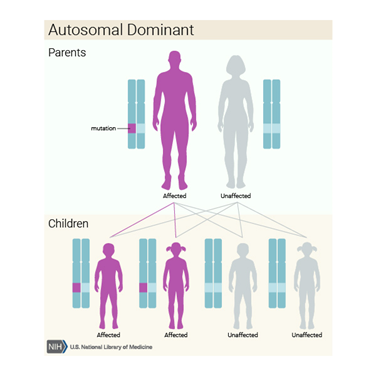
Kalıtım Paterni / Deseni
Pachyonychia congenita otozomal dominant bir durum
olarak kabul edilir, bu da her hücrede
değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu
anlamına gelir. Tüm vakaların yaklaşık yüzde 60 ila 70’inde, etkilenen bir kişi
etkilenen bir ebeveyni mutasyonu miras alır. Vakaların yüzde otuz ila 40’ı yeni
(de novo) mutasyondan kaynaklanmaktadır.üreme hücrelerinin (yumurta veya sperm)
oluşumu sırasında veya erken embriyonik gelişiminde ortaya çıkan gende. Bu
vakalar ailelerinde düzensizlik öyküsü olmayan kişilerde görülür.
Patojenik varyant etkilenen bir ailede tanımlanmışsa, yüksek riskli bir hamilelik için doğum öncesi test
mümkündür.
Teşhis Yöntemleri ve Tedavileri
Pakyonychia konjenita (PC) için klinik tanı kriterleri arasında, genetik olarak on
yaşına kadar PC ile teyit edilmiş bireylerin% 97’sinde mevcut olan ayak tırnağı
kalınlaşması, plantar keratoderma ve plantar ağrısı üçlüdür.
Pachyonychia congenita (PC) , aşağıdaki klinik özelliklere ve / veya aile öyküsü bulgularına sahip
kişilerden şüphelenilmelidir .
Klinik özellikler
– Altta yatan kabarcıklarla birlikte nasır içeren
Plantar keratoderma
– Plantar ağrısı
– Ayak tırnaklarına veya birkaç ayak tırnaklarına veya
tırnaklara sınırlandırılabilen hipertrofik tırnak distrofisi (Bkz. Şekil 1 ve
Tablo 2 )
– Yaygın steatositomalar / steatosistleri (iyi huylu
lezyonlar) ve genellikle ergenlikte gelişen ve erişkinlik döneminde devam eden
vellus kıl kistlerini içeren pilosebasöz kistler
– Oral lökokeratoz
– Gövde ve ekstremitelerdeki foliküler keratozlar
genellikle erken çocukluk döneminde görülür
– Palmoplantar hiperhidrozu (<% 50)
– Doğumsal veya doğum öncesi dişler (yani doğumda veya bazı
etkilenen bireylerde 1 aylık )
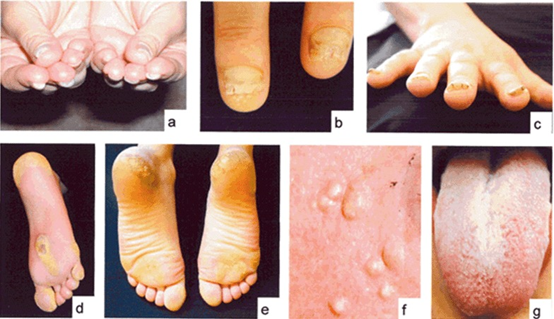
Pakyonychia congenita’nın sık rastlanan bulguları şunlardır: kalınlaşmış ve distrofik tırnaklar (hem tırnak hem de ayak tırnakları) (ac); bül (genellikle topukluların ve tabanların basınç noktalarında); hiperkeratoz (de); kistler (f); ve oral lökokeratoz (g).
Exom dizilimi ve genom
dizilimi dahil daha kapsamlı genomik testler (mevcut olduğunda) düşünülebilir. Bu testler daha önce düşünülmeyen bir teşhis sağlayabilir
veya önerebilir (örneğin, benzer bir klinik sunumla sonuçlanan farklı bir gen
veya genlerin mutasyonu).
Mevcut tedavi yöntemleritemel olarak semptomatik ağrının giderilmesine, hiperkeratotik alanların eşleştirilmesi, belirtildiği zaman ikincil enfeksiyonun tedavisi ve tekerlekli sandalye, koltuk değneği ve bastonlar da dahil olmak üzere çeşitli yürüme yardımcılarının kullanılması gibi hijyenik tımar uygulamalarına odaklanmaktadır.
Palmoplantar keratoderması. Ayakların sık sık tımar edilmesi esastır ve hiperkeratotik bölgelerin ayrıştırılmasını içerir. Ancak, çok agresif bir şekilde kesmek ağrıyı büyük ölçüde artırabilir. Bazıları soyulmadan önce ayakların ıslanmasına yardımcı olur. Enfeksiyondan kaçınmak için cildin yüzeyi ve kullanılan aletler temiz olmalıdır. Kabarcıklar steril bir iğne ile delinmeli, akışkan boşaltılmalı ve blister tavan kuruyana ve dökülünceye kadar yerinde bırakılmalıdır.
Hiperkeratozu
çıkarmak için topikal tedaviler:
* Yumuşatıcılar , örneğin
vazelin olarak ® veya lanolin içeren ürünler sık sık kullanılır. Not: Üre,
laktik asit, salisilik asit veya propilen glikol gibi keratolitikler içeren
kremler ve losyonların, bazı olumsuz yan etkileri rapor etmesiyle çok az etkisi
vardır. Tıkayıcı merhemler genellikle kötü tolere edilir.
* Oral retinoidler,
keratodermayı azaltırken, cildin altta yatan kabarma ve kırılganlığını
etkilemez ve bazen ağrıyı artırır. Dozun dikkatlice düzenlenmesi gereklidir [
Gruber et al 2012 ].
* Özel ortezler veya iç
tabanlar, fitil çoraplar, havalandırılmış ayakkabılar veya yastıklı ayakkabılar
ağrının azalmasına yardımcı olabilir, ancak ağrı günden güne değişebilir ve
bazen de dinlenme sırasında şiddetli olabilir.
İdeal
vücut ağırlığını korumak, hiperkeratoz ve ağrıyı azaltmada bir faktör olabilir.
Yürümeyi veya ayakta durmayı sınırlamak travmayı azaltmaya yardımcı olabilir ve
sonuçta ortaya çıkan kabarcıkları, nasır ve ağrıları hafifçe azaltabilir.
PC’li
bireylerde plantar ağrının kökeni, doğası ve altında yatan mekanizma yeterince
anlaşılmamıştır. Son zamanlarda yapılan birkaç çalışma, nöropatik ağrı
tedavilerinin plantar ağrı yaşayan PC’li bireylerde faydalı olabileceğini
düşündürmektedir [ Pan et al 2016 ; Wallis ve diğerleri 2016 ]
Tırnak distrofisi.Kalınlaşmış tırnaklar tipik olarak ağrılı değildir, ancak enfekte olduğunda veya travmatize edildiğinde böyle olur. Çok kalın tırnaklar ve çocuklar için etkili bir araç, tırnaklara basınç uygulamamış olan giyotin tipi bir hayvan tırnak makasıdır. Sık kullanılan diğer aletler, tıraş bıçağı veya cerrahi bıçaklar veya Dremel ® aleti gibi zımparalardır .
Bakteriyel
veya fungal enfeksiyonlar meydana gelirse, sistemik antibiyotikler veya
antifungaller gösterilir.
Özellikle
sorunlu tırnaklar cerrahi olarak başarıyla çıkarılabilir.
Oral lökokeratoz.İyi ağız hijyeni ve sık sık diş fırçasıyla fırçalama, dilde ve ağız mukozasında kalın, beyaz lekelerin görünümünü önemli ölçüde artırabilir; Bununla birlikte, çok kuvvetli bir şekilde yapılırsa, fırçalama ayrıca reaktif hiperkeratoz ile sonuçlanan mukozayı travmatize edebilir.
Bazı
kişiler oral antibiyotiklere cevap olarak lökokeratoz azaldığını ve olası bir
bakteriyel katkı olduğunu bildirmişlerdir; daha muhtemel gelişme,
antibiyotiklerin anti-enflamatuar özelliklerine bir cevap olabilir.
Foliküler hiperkeratoz. Özellikle çocuklar ve gençler için can sıkıcı olan bu bulgu alfa-hidroksi asit kremleri veya losyonları veya keratolitik yumuşatıcılarla tedavi edilebilir; ancak, bu tedaviler PC için özellikle etkili olmayabilir. Örneğin vazelin olarak yumuşatıcıların kullanılması ® veya lanolin içeren ürünler etkili olduğu rapor edilmiştir.
Laringeal tutulumlu lökokeratoz.Yalnızca PC-K6a’lı çocuklarda bildirildiği üzere, solunum yetmezliği, zaman zaman havayolunu yeniden kurmak için acil cerrahi müdahale gerektiren hayatı tehdit edebilir. Açık bir hava yolunun sürdürülmesi için gerekli cerrahi işlemler tekrarlanır; ancak ses kısıklığını iyileştirmeyi amaçlayan gırtlak cerrahi prosedürleri, durumu daha da kötüleştirme eğiliminde olabileceğinden kaçınılmalıdır.
Kistler. Steatositoma multipleks ve diğer pilosebasöz kistler, 11 numara bıçakla kesi ve kist içeriğinin müteakip ifadesiyle (“kesi ve drenaj”) tedavi edilebilir. Sekonder enfeksiyon durumunda oral antibiyotikler belirtilebilir. Enfeksiyon önemliyse kültür elde edilmelidir. İntralesyonel steroid enjeksiyonu (örneğin, triamsinolon), enfeksiyondan şüphelenilmezse alanın iltihaplanmasını azaltabilir. Gerekirse, kistler eksize edilebilir.
Gelişememek. Bebeklikteki zayıf beslemenin, gerekli emmeyi azaltmak için genişletilmiş bir açıklığa sahip yumuşak bir meme ucu kullanılarak iyileştirildiği bildirilmiştir.
Sekonder
Komplikasyonların Önlenmesi
Cilt
veya tırnakların tımar veya travma sonrası enfeksiyonu, PC’de görülen en sık
görülen ikincil komplikasyondur.
– Bakım öncesi ve sonrası
hijyen ve temiz alet kullanımı bu komplikasyonları en aza indirir.
– Enfeksiyon meydana
geldiğinde antibiyotik gösterilebilir.
– Hafif bir çamaşır suyu
çözeltisinin kullanıldığı basit bir “çamaşır suyu banyosu” rejimi
enfeksiyonların önlenmesine yardımcı olabilir.
Uluslararası Pachyonychia Congenita Araştırma
Kayıtları (IPCRR) verilerine dayanarak, pachyonychia congenita için en son sınıflandırma mutasyona uğramış gen
tarafından yapılmıştır [ McLean et al 2011 , Wilson et al 2011 ]:
* (Patojen varyantları neden olduğu PC-K6A KRT6A )
* (Patojen varyantları neden olduğu PC-K6b KRT6B )
* PC-K6c (patojen varyantları kaynaklanan KRT6C )
* PC-K16 ( KRT16’daki patojenik varyantların neden
olduğu )
* PC-K17 ( KRT17’deki patojenik değişkenlerin neden
olduğu )
Hastalığın genetik temelinin belirlenmesinden önce PC
için önerilen sınıflandırma yalnızca klinik bulgulara dayandırılmıştır.
Tarihsel olarak, PC’nin iki büyük alt tipi, ince değişken fenotipik özelliklere
dayanıyordu (temel olarak pilosebasöz kistlerin ve doğum veya doğum öncesi
dişlerin varlığı ya da yokluğu üzerine) [ Leachman ve ark 2005 , Liao ve ark
2007 ]:
* PC-1 (Jadassohn-Lewandowski sendromu)
* PC-2 (Jackson-Lawler sendromu)
PC’li kişilerin sayısındaki artışta ayrıntılı klinik
geçmişleri ve patojenik varyasyonları ile, PC-1 ve PC-2’nin eski
sınıflandırmasının PC’li bireylerin daha geniş popülasyonu için geçerli
olmadığı açıklığa kavuşturuldu.
KRT6A , KRT6B , KRT6C , KRT16 , KRT17 . Bu
GenReview’da tartışılanlar dışında hiçbir fenotipin, bu genlerdeki patojenik
varyantlarla ilişkili olduğu bilinmemektedir.
Faktör XI eksikliği , kanın pıhtılaşmasında rol alan faktör XI proteininin bir kıtlığına (eksikliğine) bağlı olarak anormal kanamaya neden olabilecek bir hastalıktır. Bu durum, faktör XI proteininin eksiklik derecesine bağlı olarak kısmi veya şiddetli olarak sınıflandırılır. Bununla birlikte, protein eksikliğinin ciddiyetine bakılmaksızın, çoğu etkilenen birey göreceli olarak hafif kanama problemlerine sahiptir.Faktör XI sendromuna sahip kişilerdeki belirtiler aynı aile içerisinde bile değişebilir.Diğer genetik ve çevresel faktörler hastalığın derecesinin belirlenmesinde etkilidir.
BELİRTİ VE SEMPTOMLAR:
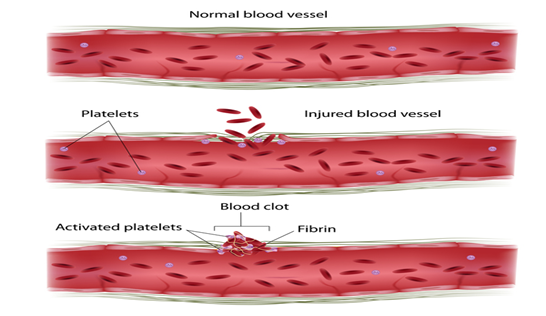
Faktör XI eksikliği vakalarının çoğu , faktör XI proteini yapmak için talimatlar veren F11 genindeki mutasyonlardan kaynaklanır . Bu protein, kan pıhtılarını oluşturan bir dizi kimyasal reaksiyon olan pıhtılaşma kaskadında rol oynar.Yaralanmaya yanıt olarak. Bir yaralanmadan sonra pıhtılar kanamayı durdurmak ve kan damarı onarımını tetiklemek için kan damarlarını kapatır.
F11 genindeki mutasyonlar , fonksiyonel faktör XI’nin eksikliğine (eksikliğine) neden olur. Bu eksiklik pıhtılaşma kademesini bozar, kanın pıhtılaşma sürecini yavaşlatır ve bu hastalıkla ilişkili kanama sorunlarına yol açar. Kalan fonksiyonel faktör XI miktarı, özel mutasyona ve F11 geninin bir veya her iki kopyasının, her hücrede mutasyona sahip olup olmamasına bağlı olarak değişir . Bununla birlikte, etkilenen bireylerde kanama problemlerinin ciddiyeti mutlaka kan dolaşımındaki faktör XI miktarına karşılık gelmez ve aynı aile içinde bile değişebilir. Diğer genetik ve çevresel faktörler muhtemelen bu durumun ciddiyetinin belirlenmesinde rol oynar.
Faktör XI eksikliğinin en sık görülen özelliği , özellikle ağız ve burun içini (travma veya burun boşlukları dahil) travma veya ameliyat sonrası uzun süreli kanamadır.) veya idrar yolu. Kanama ameliyattan sonra tedavi edilmezse, cerrahi alanda konjuge kandan (hematom) oluşan katı şişlikler gelişebilir. Bu hastalığın diğer belirti ve semptomları sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri), gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın olmayan faktör XI eksikliği onlar ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez.
Bu hastalığın diğer belirti ve semptomları sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri), gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın olmayan faktör XI eksikliği onlar ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez. Aşağıda bu hastalığı olan kişilerin sahip olabileceği belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan insanlar listelenen tüm belirtilere sahip olmayabilir. Bu tablo düzenli olarak İnsan Fenotip Ontoloji ((Human Phenotype Ontology [HPO]) isimli veri tabanı tarafından güncellenmektedir. ([https://hpo.jax.org/)
GENETİK GÖRÜLME SIKLIĞI: Faktör XI eksikliğinin dünya genelinde yaklaşık 1 milyon kişiden 1’ini etkilediği tahmin edilmektedir. Orta ve doğu Avrupa (Aşkenazi) Yahudi soyuna sahip kişilerde, bu popülasyondaki 450 kişiden 1’inde meydana gelen şiddetli bozukluk bozukluğu çok daha yaygındır. Araştırmacılar, faktör XI eksikliğinin gerçek prevalansının bildirilenden daha yüksek olabileceğini öne sürüyorlar , çünkü hastalığın hafif vakaları çoğu zaman tıbbi yardıma gelmiyor.
KALITIM PATERNİ /DESENİ: Otozomal resesif paternde ciddi faktör XI eksikliği azaldıbu, her bir hücrede F11 geninin her iki kopyasının da mutasyonlara sahip olduğu anlamına gelir . Bu bireylerin ebeveynlerinin her biri, mutasyona uğramış genin bir kopyasını taşır ve kısmi faktör XI eksikliğine sahiptir ; nadiren durumun ciddi belirtileri ve semptomlarını gösterirler.
Bazı ailelerde bu durum otozomal dominant paternde kalıtsaldırbu, her hücrede değiştirilmiş F11 geninin bir kopyasının , bozukluğa neden olmak için yeterli olduğu anlamına gelir . Bu gibi durumlarda, etkilenen bir kişinin durumu olan bir ebeveyni vardır. Kazanılan faktör XI eksikliği formu kalıtsal değildir ve ailelerde çalışmaz.
Teşhis Yöntemleri ve Tedaviler: Faktör XI eksikliği sendromunun tanısı için 3 önemli belirti şu şekildedir: 1)Ağız ve burun içi travma veya ameliyat sonrası uzun süreli kanama. 2)Sık sık burun kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanaması. 3)Bu hastalığa sahip olan kadınlarda ağır veya uzun süreli adet kanaması (menoraji) ve doğumdan sonra uzun süreli kanama. Ayrıca faktör XI eksikliği sendromu için bazı testler yapılmaktadır. Ancak hastalık tanısı için gerekli değildir.
1)GENETİK TEST: Genetik test, kromozom, gen veya proteinlerdeki değişiklikleri tanımlayan bir tıbbi test türüdür. Genetik testin sonuçları, şüpheli bir genetik durumu onaylayabilir veya ekarte edebilir veya bir kişinin genetik hastalık geliştirme veya geçme şansını belirlemeye yardımcı olabilir. Şu anda 1000’den fazla genetik test kullanılıyor ve daha fazlası geliştiriliyor. Genetik test için çeşitli yöntemler kullanılabilir: Moleküler genetik testler (veya gen testleri), genetik bir bozukluğa yol açan varyasyonları veya mutasyonları tanımlamak için tekli genleri veya kısa DNA uzunluklarını inceler. Kromozomal genetik testler, genetik bir duruma neden olan bir kromozomun ekstra bir kopyası gibi büyük genetik değişikliklerin olup olmadığını görmek için bütün kromozomları veya uzun DNA uzunluklarını analiz eder. Biyokimyasal genetik testler, proteinlerin miktarını veya aktivite seviyesini inceler; herhangi birindeki anormallikler, genetik bir bozuklukla sonuçlanan DNA’daki değişiklikleri gösterebilir. Genetik test isteğe bağlıdır. Testin sınırlamalar ve risklerin yanı sıra faydaları olduğu için, test edilip edilmeyeceği konusundaki karar kişisel ve karmaşık bir karardır. Bir genetikçi veya genetik danışman, testin artıları ve eksileri hakkında bilgi vererek ve testin sosyal ve duygusal yönlerini tartışarak yardımcı olabilir.
2)KISMİ TROMBOPLASTİN ZAMANI (PTT): Kısmi tromboplastin zamanı (PTT) kanın pıhtılaşmasının ne kadar sürdüğünü gösteren bir kan testidir. Kanama probleminiz olup olmadığını veya kanınızın pıhtılaşmaması durumunda size yardımcı olabilir.
3)PROTROMBİN ZAMANI (PT): İlgili bir kan testidir. HASTALIĞIN DİĞER İSİMLERİ: F11 eksikliği faktör 11 eksikliği hemofili C hemofili C plazma tromboplastin öncül eksikliği PTA eksikliği Rosenthal faktörü eksikliği Rosenthal sendromu Rosenthal hastalığı
Faktör XI Eksikliği İçin Yurt Dışındaki Kuruluşlar: 1) Amerika Hemofili Federasyonu 2) Ulusal Hemofili Vakfı 3) Kanada Hemofili Derneği 4) Genetik ve Nadir Hastalıklar (GARD) Bilgi Merkezi 5) Dünya Hemofili Federasyonu
Genel
Bilgi, Genetik Değişiklikler/Etken Faktörler
Pallister-Hall sendromu (PHS), vücudun birçok
bölümünün gelişimini etkileyen genetik bir hastalıktır. Ortak özellikler
arasında ekstra parmaklar ve / veya ayak parmakları (polidaktil), parmaklar
veya ayak parmakları arasında ekstra cilt (sindaktil), beyinde hipotalamik
hamartom denilen bir anormal büyüme ve iki üçlü epiglotis olarak bilinen hava
yolunun bir malformasyonu bulunur. Nadir durumlarda bifid epiglot, solunum
yetmezliğine yol açabilir. Çoğu durumda hipotalamik hamartom sorun yaratmasa
da, bazı durumlarda nöbetler, büyüme hormonu eksikliği, erken ergenlik veya
kortizol eksikliğine neden olabilecek birçok hormonun (panhipopitüitarizm)
eksikliği gibi nörolojik sorunlara neden olabilir. PHS’nin diğer semptomları
arasında deliksiz anüs, böbrek anomalileri, kalp defektleri, küçük genital
organlar, parmak eksikliği, tırnak problemleri, yarık damak, bifid uvula ve
gelişim gecikmesi ve davranış problemleri sayılabilir. GLI3 genindeki
mutasyonlar Pallister-Hall sendromuna neden olur. Bu gen, genlerin belirli
hücrelerde açılıp kapatılmayacağını düzenleyen bir işlem olan gen ekspresyonunu
kontrol eden bir protein yapmak için talimatlar sağlar. Gelişim sırasında
belirli zamanlarda belirli genlerle etkileşime girerek, GLI3 proteini doğumdan
önce birçok organ ve dokunun normal şekillenmesinde (desenlenmesinde) rol
oynar.
Pallister-Hall sendromuna neden olan
mutasyonlar tipik olarak GLI3 proteininin anormal derecede kısa bir
versiyonunun üretilmesine yol açar. Hedef genleri açıp kapatabilen normal GLI3
proteininin aksine, kısa protein sadece hedef genleri kapatabilir
(bastırabilir). Araştırmacılar, protein işlevindeki bu değişikliğin erken
gelişimi nasıl etkilediğini belirlemek için çalışmaktadır. GLI3 mutasyonlarının
polidaktil, hipotalamik hamartom ve Pallister-Hall sendromunun diğer
özelliklerine neden olabileceği kesin değildir.
Belirti ve
Semptomlar
PHS’li çoğu hasta doğumda üçüncü veya dördüncü
dereceden iskelet polidaktili veya postaksiyal polidaktili görülürken,
beraberinde her ikisine de kütanöz sindaktili ve tırnak displazisi eşlik
edebilir. Yüz özellikleri kısa burun, düz burun köprüsü ve alçak ayarlanmış,
arka acılı kulakları içerebilir. Bazı hastalarda yarık damak, yarık uvula ve
multipl bukal frenula bildirilmiştir. Asemptomatik bir bifid epiglotis
neredeyse patognomoniktir, ancak bazı hastalarda potansiyel olarak ölümcül
solunum yetmezliğine yol açan daha ciddi posterior laringeal yarıklar görülür.
Hipotalamik hamartom genellikle asemptomatiktir, ancak panhipopituitarizm ile
ilişkili olabilir. Akut primer adrenal yetmezlik ciddi vakaların yani sıra,
daha hafif adrenal yetmezlik formlarında da görülebilir. Erken ergenlik bazı
durumlarda kendini gösterir. Nörolojik tutulum, gelastik epilepsiyi (yüz
ekşitmeden, gülüşmekten veya kahkaha olarak ortaya çıkan nöbetler) veya diğer
nöbet tiplerini içerebilir. Vajinal atrezi veya hidrometrokolpos, microphallus
veya kriptorşidizm de dahil olmak üzere böbrek agenezisi veya displazisi ile
diğer genitoüriner anomaliler bildirilmiştir. Diğer bulgular arasında
intrauterin gelişme geriliği, anormal akciğer lobasyonu, mezomelik kısalması
ile genelleşmiş iskelet displazisi ve uzuvların radyal yaylanması, imperforat
anüs ve konjenital kalp defektleri sayılabilir.
Genetik Görülme Sıklığı
Bu durum çok nadirdir; prevalansı
bilinmemektedir. Bugüne kadar yaklaşık 100 hasta bildirilmiştir.
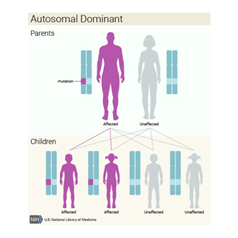
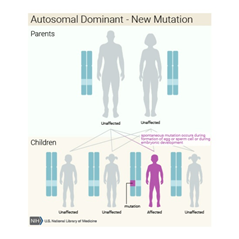
Kalıtım
Paterni/Deseni
Bu durum otozomal dominant paternde kalıtsaldır, bu da her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir. Bazı durumlarda, etkilenen bir kişi, etkilenen bir ebeveynden GLI3 geninde bir mutasyon devralır. Diğer vakalar, gendeki yeni mutasyonlardan kaynaklanır ve ailelerinde hastalık öyküsü olmayan kişilerde ortaya çıkar.
Iafolla ve diğerleri (1989) manyetik rezonans
görüntülemenin en değerli tanı aracı olduğuna işaret etmişlerdir; BT
taramasının tümörü kaçırdığı bildirildi.
Pallister-Hall sendromu üzerine uluslararası
bir atölye çalışması (Biesecker ve diğerleri, 1996) bu varlık için minimal tanı
kriterleri geliştirmiştir. Bir ailedeki indeks olgusunda, tanı kriterlerini
karşılamak için hem hipotalamik hamartom hem de merkezi polidaktili olmalıdır.
İndeks olgunun birinci derece akrabalarında hipotalamik hamartom veya
polidaktili (merkezi veya postaksiyal) bulunmalı ve otozomal dominant paternde
veya gonadal mozaikliği ile uyumlu bir şekilde kalıtım gösterilmelidir. Şüpheli
olguların klinik değerlendirmesi için öneriler sunuldu. Biesecker ve diğerleri
(1996), hipotalamik hamartomun PHS’ye özgü olmadığı sonucuna varmıştır.
Hastalıkla İlişkili Genler
Pallister-Hall sendromuna GLI3 genindeki
mutasyonlar neden olur. Kalıtım otozomal dominanttır, ancak vakaların yaklaşık
dörtte birinde Pallister Hall sendromu yeni (de novo) bir mutasyondan
kaynaklanır
Q
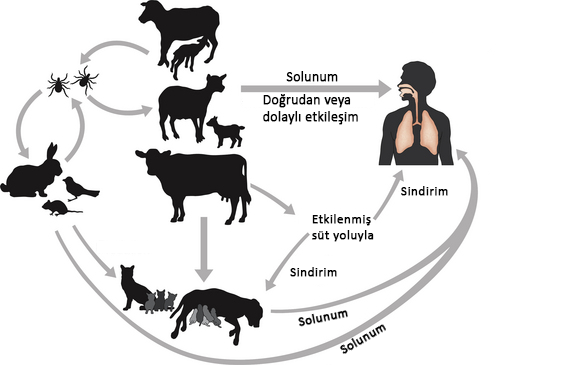
ateşi, Coxiella Burnetii bakterisinden kaynaklanan bir
hastalıktır. Genellikle keçi, koyun gibi hayvanlarda doğal olarak gözlemlenen
bu bakteri, solunum veya bakteriden etkilenmiş yemekleri yiyip içerek insanlara
geçebilir.
Etken Faktörler
Coxiella Burnetii bakterisinin bulaştığı hayvanlar; vucüt sıvılarıyla (doğum sırasında, dışkısıyla, sütüyle) veya hayvanın etinin yenmesiyle bakteriyi bulaştırabilir. Keneler bu hastalığın yayılmasında önemli faktörlerdir. Etkilenen hayvanlarla etkileşime geçen insanların hastalığa yakalanma ihtimali yüksektir.
Q
ateşi hastalığı genetik değildir. Bakteriyel bir hastalıktır.
Belirti ve Semptomlar
Q
ateşi hastalığının belirtileri kişiden kişiye değişiklik gösterebilir.
Hastalık, semptomsuz olabilir, aküt veya kronik olabilir.
Aküt
Q hastalığında, bakteriye maruz kalındıktan sonra 2-3 hafta içerisinde semptomlar
ortaya çıkar. Semptomlar arasında; ateş, yorgunluk, kas
ağrıları, mide bulanması, kusma, göğüs ve karın ağrısı, üşüme, terleme, ishal,
kilo kaybı ve kuru öksürük olabilir.
Hamilelerde düşük,
ölü doğum, erken doğum ve bebeğin gereğinden az kiloda doğması olabilir.
Ağır vakalarda ise
akciğer veya karaciğerde iltihaplanma görülebilir.
Kronik Q
hastalığı, bakterinin vucüda girmesinden aylar veya yıllar sonra belirti gösterebilir.
Genellikle, kalp ve damar hastalığı olan veya bağışıklık sistemi baskılanmış
hastalarda görülür. Görülme sıklığı %5ten azdır. Semptomları en çok
etkiledikleri bölgeye göre değişir, belirli bir semptomu yoktur. Uzun süreli
yorgunluk, ateş hali, eklem ağrılarına sebep olabilir.
Teşhis Yöntemleri ve Tedaviler
Q
hastalığının belirtileri genel olduğu için, kan tahliliyle tetkik edilmesi
gerekir. Özellikle hayvanlarla temasta olan hastalara, doktor kan tahlilinin
sonucunu beklemeden tedaviye başlayabilir. Antibiyotik genellikle ilk tercih
edilen tedavi şeklidir.
Aküt
Q hastalığı olan hastalarda, ilk üç günde antibiyotiğe başlamak en verimli
olur. Antibiyotiğin cevap vermediği durumlarda anti inflamatuvar ilaçlar
kullanılabilir.
Kronik
Q hastalığının tedavisi daha zordur, en çok etkilenen bölgeye göre kişiden
kişiye değişiklik gösterir. Bazı durumlarda ameliyata başvurulması gerekli
olabilir.
Akondroplazi, kısa ekstremiteli (uzuvlu)
cüceliktir. Akondroplazi, uzun kemiklerin kıkırdak içindeki kemik dokuya
dönüşmesini (osifikasyon) engelleyen bir rahatsızlıktır. Bu rahatsızlığa FGFR3
geninde yerleşik mutasyon (değişim) sebep olur. Hastaların% 80’inde
kendiliğinden (spontan) mutasyon görülken geride kalan% 20’inde hastalık
otozomal dominant olarak kalıtılır.
Belirti ve Özetler
Akondroplazi hastalığında belirtileri; kısa oğlan,
alışılmamış şekli büyük kafa (makrosefali), çıkıntılı alın, basık burun, kısa
kol ve uzakta, birinden uzaklanırken karın ve doğlar ve kısa ellerdir.
Akondroplazi hastası yeni doğanlarda kubbeli olduğu,
geniş alındığı görülür. Hidrosefali görülebilir. Bebeklik döneminde hipotoni,
akondroplazinin tipik bir şekildedir.
Genetik Görülme Sıklığı
Akondroplazi kadın ve erkeklerde eşit oranda görülür.
15.000-35.000 doğumda bir görülüyor.
Bazı popülasyonlarda, akondroplazinin görülme sıklığı
daha fazladır. Yıllar, Danimarka’da 6400 doğumda yaklaşık 1 vakada ve Latin
Amerika’da 10.000 doğumda yaklaşık 1 vakada meydana geldiği tahmin
edilmektedir. Ancak bir ırkın daha sık etkilendiği belgelenmemiş.
Kalıtım Paterni/ Deseni
Akondroplazi otozomal dominant olarak kalıtılır. Yani
hastalık temizleme için, genin bir ödenmelidir.
Teşhis Yöntemleri ve Tedaviler
Teşhis Yöntemleri
Akondroplazinin klinik ve radyolojik özellikleri iyi
durumda. Doğumdan hemen sonra klinik ve radyolojik olarak tanınabilir. Tipik
bulgulara sahip olanlardan birini tanıyı doğrulamak için moleküler genetik
testlere ihtiyaç duymazlar. Yeni doğanlarda tanıya dair bir şüphe uyandığında,
tanıyı doğrulamak için X-ışını (radyografi). FGFR3 geninde mutasyon var olup
olmadığına moleküler genetik testlerle bakılır.
Akondroplazi tanısında kullandığı klinik bulgular;
• Orantısız Kısa Erkek
• Makrosefali
• Orta yüz hipoplazisi ile tasarı dismorfik yüz
görünümü
• Dirsek ekstensiyonunda rekabet
• Kısa el ve ayak parmakları
• Lombar lordoz
• Çarpık yönetimi
Tedaviler
Bebeklerde foramen magnumun dekompresyonu ve
hidrosefali için şant gerekebilir.
Kulak tedavisi ve seröz otitis media tedavisi, işitme
problemlerinin yapılması ile birlikte. Konuşma terapisi önerilebilir. Çarpık
temizleme cerrahi ile düzeltilebilir. Lomber laminektomi gerekebilir.
Çocuklukta kilo alımı, daha sonra oluşabilecek komplikasyonları önlemek için
kontrol edilebilir. Boyundaki cini incitebilir aktivitelerden kaçınılabilir.
Sosyal ve psikolojik destek önerilebilir.
• Hidrosefalı: semptomların (hızlı kafa büyümesi,
görüşmede, baş ağrısı, şişkin alınabilir) ortaya çıkması, semptomlar (kafa
kafalarının büyümesi, kafa kafalarının büyümesi).
• Boyun ekleminde (kraniyoservikal bölge) daralma:
Ense dekompresyonu gerekebilir.
• Obstrüktif uyku apnesi: Kilo verme, bademcikleri ve
adenoidleri kapatma ameliyatı (adenotonsillektomi), pozitif hava yolu ve
nadiren boyun açıklığı için işleme (ameliyat) işlemi yapılabilir.
• Orta kulakta fonksiyon bozukluğu: Sıkı olabilir orta
kulak iltihabını ve olası işitme kaybını engellemek için, 7-8 yaşına kadar
kulak tüpleri vardır.
• Kısa oğlan: Büyüme hormonu kullanımı üzerine yapılan
araştırmalar, büyümenin başlangıçtaki hızılanmasını, ancak zamanla etkisi
azalır ve kalıcı faydalanabiliyor.
• Çarpık bacak: Ortopediste görünmek gerekir.
• Omurga Uzaklığı: Yaşamın ilk 12-18 liman
desteklenmemiş oturma yasağı dahil olmak üzere önleyici tedbirler, omurga
bölgesi (kifoz) sabit bir yerde eğri odada riskini içerir. Ciddiyet derecesine
bağlı olarak, bantlama veya cerrahi gerekebilir.
Hastalıkla İlişkili Genler
Bu hastalığa FGFR3 (fibroblast büyüme faktörü 3) adlı
gende oluşan mutasyon sebepleri olur. Bu gen, beyin ve kemik dokusunun
gelişimiinde görev alan bir proteini kodlar. Araştırmacılar, bu mutasyonların
FGFR3 proteinlerinin aşırı aktif olması nedeniyle neden olabilir, bunun için
iskelet gelişimini engellediğini ve bu rahatsızlıklara bakarken kemik
oluşumesinde rahatsızlıklara yol açtığını gösteriyor.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Galaktozemi, bireyin basit bir karbonhidrat
olan galaktozu bir başka karbonhidrat olan glikoza çevirmesini etkiler.
Özellikle süt ve süt ürünlerinde bulunan laktozun içerisinde, glikoz ve
galaktoz bulunur. Laktozlu ürün tüketilerek alınan galaktozun vucütta
kullanılabilmesi için glikoza çevirilmesi gerekir. Klasik galaktozemide bu çevirimi yapan GALT
(galactose-1-phosphate uridylyl transferase) enzimi eksikliği hastalığa sebep
olur. Otozomal çekinik olan, kalıtımsal bir hastalıktır.
Belirti ve Semptomlar
Galaktozemi, birden fazla alt gruba
ayrılmıştır.
Klasik galaktozemi, hastalığın en yaygın ve
ağır şeklidir. Tip 1 olarak da bilinir. Yeni doğanlarda düşük galaktozlu diyet
hemen kullanılmazsa birkaç gün içerisinde komplikasyonlar meydana gelebilir.
Genellikle beslenme güçlüğü, kilo almada sorunlar, sarılık, karaciğerde büyüme,
kusma ve karında şişkinlik gözükür. Sonrasında ise ciddi bakteriyel
enfeksiyonlarla karşılaşılabilir. Hasta bireylerde katarakt, geç büyüme ve kız
çocuklarında yumurtalıkların erken fonksiyon kaybına yol açabilir.
Galaktozemi tip 2 ve tip 3 klasik tipten farklı
semptomlara yol açar ve görülme sıklıkları değişiklik gösterir.
Galaktozemi tip 2, galaktokinaz enzimi
eksikliğinden dolayı ortaya çıkar. Klasik tipe göre daha az saylık sorunu
yaşanır. Katarakt gözükebilir fakat uzun sğreli etkileri beklenmez.
Galaktozemi tip 3, galaktoz epimeraz eksikliği
olarak da geçer. Semptomların şiddeti değişiklik gösterebilir. Katarakt,
büyümede gecikme, zihinsel engel, karaciğer ve böbrek sorunlarına sebep
olabilir.
Genel olarak; galaktozun glikoza
çevirilmemesinden kaynaklanan vucüttaki fazla galaktoz, birikime sebep
olabilir. Bu birikim de karaciğerde büyümeye, siroz başlangıcına, karında asit
birikmesine, böbreklerde ve beyinde ciddi hasarlara sebep olabilir.
Genetik Görülme Sıklığı
Tip 1 Galaktozemi, 30.000-60.000 de 1
yenidoğanda görülebilir.
Tip 2 100.000de 1 den daha az görülürken tip 3
galaktozemi oldukça nadirdir.
Kalıtım Paterni/Deseni
Galaktozemi, otozomal çekinik bir hastalıktır.
Yani, iki genetik ebeveynden de hastalık geninin gelmesi gerekir. Aile
geçmişinde bu hastalık varsa gerekli genetik testler yapılmalı ve mutlaka önlem
alınmalıdır. Ayrıca, akraba evliliklerinde hastalığın görülme olasılığı
artacağından akraba evliliklerinden kaçınılmalıdır.
Teşhis Yöntemleri ve Tedaviler
Yenidoğanlarda topuktan alınan kan testinde galaktozemi
neredeyse %100 oranında belirlenebilmektedir. Yetkili doktor tarafından
galaktozemi teşhisi konulması gerekir.
Laktoz toleransı testi bu hastalara yapılmamalıdır.
Hastalığı tamamen ortadan kaldıracak bir tedavi yöntemi
şimdilik bilinmemektedir. Bu hastalığa uygun beslenme düzeniyle semptomlar en
az seviyeye çekilebilir, hatta kaldırılabilir. Beslenme düzeninden laktoz ve
galaktoz bulunan tüm ürünler çıkartılmalıdır. Hayatı boyunca bu bireyin
beslenme düzeninin laktozsuz olacağının kabullenilmesi gerekir.
Hastalıkla İlişkili Genler
Klasik galaktozemi, GALT geni sayesinde
üretilen GALT enziminin yokluğu veya düzgün çalışmamasından ortaya çıkan bir
hastalıktır.
İyonize radyasyona maruz kalma sonucu akut, gecikmiş olarak
veya kronik olarak etkilerini gösteren bir hastalıktır. Büyük dozlarda etkiler
hemen ortaya çıkarken, küçük dozlarda radyasyonun vücutta birikmesi sonucu
genetik ve uzun süreli etkiler görülmektedir. Radyasyondan zarar görüp hasara
uğrayan hücrelerin tamiri için tedavi bulunmamaktadır. Ancak yakın zamanda FDA (Amerikan
Gıda ve İlaç Dairesi), radyoaktif elementlerin vücuttan atılmasında etkili olan
ilaçların onayını vermiştir. Hücre hasarı geri dönüşsüzdür ancak bu ilaçlarla
hastalarda radyasyona maruz kalma sonucu çıkan belirtiler tedavi
edilebilmektedir.
İlk Radyasyon Hastalığı vakaları Hiroshima ve Nagasaki
nükleer patlamalarından sonra görülmüş, Japon hekimler bu hastalığı “gözle
görülür hiçbir hasar olmadan, birden ortaya çıkan hastalık” olarak
tanımlamıştır. Günümüzde, bu hastaların radyasyon hastalığına yakalandığı anlaşılmıştır.
Düşük miktarda maruziyet sonucu bu hastalık soğuk algınlığında görülen hafif
belirtilerle hastayı terk edebiliyorken; yüksek miktarda maruziyet Chernobyl
patlamasında olduğu gibi hastada ölümcül etkiler yaratabilir.
Toplam doz ve doz oranı radyasyonun somatik ve genetik
etkilerini belirler. Radyasyon dozu belirtilirken üç ölçü birimi kullanılır:
Roentgen, rad ve rem. Roentgen (R), havadaki x ve gamma ışınlarının miktarını
belirtirken; rad (radiation absorbed dose), tüm radyasyon tiplerinde absorbe
edilen enerji miktarı için kullanılır. Rem ise nötronlar gibi bazı radyasyon
türlerinin, eşdeğer miktarda emilen enerji için ne kadar biyolojik etki
üretebileceğini gözlemlemek için kullanılır.
Hastalığın Diğer İsimleri
null
Radiation Disease
Radiation Effects
Radiation Illness
Radiation Injuries
Radiation Reaction
Radiation Syndrome
Belirti ve Semptomlar
Akut radyasyon hastalığı bulantı, kusma, ishal, anoreksiya,
baş ağrısı, halsizlik ve taşikardi (kalp çarpıntısı) belirtileriyle kendini
gösterir. Hafif olgularda belirtiler birkaç saat veya gün içinde kaybolur. Akut
radyasyon hastalığının yüksek doz veya düşük doza bağlı olarak ortaya çıkan çeşitli
tipleri vardır.
Akut radyasyon hastalığı çeşitleri doz, doz oranı, maruz
kalan vücut bölümü ve maruziyetten sonra vücuttan atılması için geçen süreye
göre hastada gözlemlenmektedir. Radyasyona
maruziyet sonucu dakikalar içinde radyasyon tüm vücudu etkiler.
Akut Radyasyon hastalığı üç aşamayla gelişir: İlk aşama
birkaç dakikadan birkaç güne kadar sürebilir ve hastada mide bulantısı, diyare
ve kusma görülür. Bundan sonraki aşamada hasta birkaç hafta iyileşme
belirtileri gösterir. Son aşamada ise hastadan hastaya değişmekle beraber
kardiyovasküler, gastrointestinal, hematopoietik veya sinir sistemi
rahatsızlıkları görülmektedir. Yüksek dozda (3000 rad’dan fazla radyasyon
maruziyet) birkaç saat içinde ölümcül kardiyovasküler belirtiler ve sinir
sistemi rahatsızlıkları, mide bulantısı, kusma, anksiyete (endişe), konfüzyon
(bilinç bulanıklığı) ve bilinç kaybı görülmektedir. 5-6 saat sonunda tremor
(titreme) ve konvülziyon (havale) başlar ve üç gün sonunda ölüme sebep olur. 400
rad’dan fazla radyasyona maruz kalma sonucu mide bulantısı, kusma, elektrolit
dengesizliği, diyare (ishal), sıvı kaybı, plazma hacminde azalma gibi
gastrointestinal rahatsızlıklar ortaya çıkar. 200-1000 rad arasında, 6 ila 12
saatlik radyasyon maruziyeti sonucunda ise anoreksiya, ateş, halsizlikle
seyreden hematopoietik rahatsızlıklar oluşur.
Nedenleri
İyonize radyasyonun en büyük kaynağı özellikle kanser tedavisinde kullanılan yüksek enerjili x-ray ışınları ve radyum vb. radyoaktif elementlerdir. Bunun dışında nükleer santrallerde meydana gelen patlamalar veya nükleer silah savaşları da başlıca hastalık kaynaklarıdır. Örneğin, Hiroshima, Nagasaki ve Chernobyl nükleer santrallerinde patlamalar sonucunda bu bölgeler ve çevre bölgelerdeki insanlarda kanser, mutasyon ve genetik hasarlar meydana geldi; uzun yıllar boyunca radyasyon maruziyeti ve etkileri devam etti.
Vücudun radyasyona maruz kalan kısmı bu hastalığın ortaya
çıkmasında önemli bir faktördür. İnsan vücudu 200 rad’a kadar radyasyonu
ölümcül risk olmadan absorbe edebilmektedir. Radyasyon dozu 450 rad’a
ulaştığında ölüm riski %50’ye ulaşırken bazen 600 rad kısa sürede ölüme sebep
olabilir.
Radyasyonun vücutta dağılımı da hastalığın seyrini etkileyen
başka bir faktördür: Örneğin bağırsak ve kemik iliğinin korunması hastanın
ölümünü engeller.
Etkilediği popülasyon
Radyasyon hastalığı kadın ve erkekleri eşit oranda
etkilemektedir.
Teşhis
Teşhis, radyasyon maruziyeti öyküsüne bağlı olarak
belirlenir. Maruz kalma ve kusma arasında geçen süre maruziyet seviyesinin
tespitinde önem teşkil eder.
Maruz kalan hastaların Geiger sayacı veya tüm vücut analizi
için kullanılan sayaçlarla izlenmesi gerekmektedir.
Tedavi
Radyoaktif elementlerin deriden maruziyeti sonucu derhal
kontaminasyon (uzaklaştırma) sağlanmalıdır. Hasta bol sıvıyla durulanmalı ve
EDTA (radyoaktif izotopların etkisini indirgemek için kullanılan bir madde) vb.
maddeler uygulanmalıdır. Ayrıca küçük yaralar ve maruz kalan dokular da
kontaminasyonu engellemek için iyice temizlenmelidir. Eğer radyasyon içeren bir
madde ağız yoluyla vücuda alınırsa kusturma veya mide yıkaması
yapılmalıdır.
2015 yılında radyasyonun kemik iliğine sebep olduğu yetişkin
ve çocuklarda kullanılmak üzere Amgen firması tarafından Neupogen adlı bir ilaç
piyasaya sürüldü. Bunun haricinde radyoaktif talyum maruziyetinin tedavisinde,
FDA onaylı aslında endüstride kullanılan Prusya mavisi adında bir pigment
kullanılmaktadır. Vücutta bu elementlerin absorblanmasını engelleyerek etki
göstermektedir. Plütonyum, amerikyum ve bakır radyoaktiflerine karşı Ca-DTPA ve
Zn-DTPA kullanılması FDA tarafından onaylanmıştır.
Genel Bilgi, Genetik
Değişiklikler/Etken Faktörler
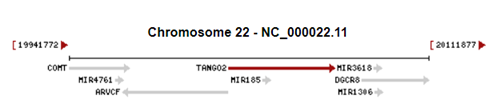
TANGO2, 22. Kromozomda (22q11.21) bulunan
protein kodlayan bir gendir. Bu gendeki mutasyon, otozomal çekinik olarak
gelecek nesle aktarılır. Etkilenen bireyler, metabolik kriz de denilebilen
aralıklı akut rahatsızlıklara yakalanabilir. Uzun süre açlık veya nexle gibi
bir başka bu krizleri tetikleyebilir. Metabolik krizler sırasında aritmiler
(kalp atışı ritminin anormal değişimi), rabdomiyoliz (iskelet kası dokusundaki
hasar sebebiyle bozulma) veya
ensefalopati (beyinde oluşan hasar, arıza) meydana gelebilir. Her birey bu
hastalığı farklı deneyimleyebilir.
Bilinen bir tedavisi yoktur
fakat araştırmalar devam etmektedir.Hastalığın etkilerini en aza indirgemek ve kontrol
altında tutmak şimdilik asıl amaçtır.
Görülme sıklığı
Bu hastalık literature 2016 yılında girmiştir. TANGO2
Research Foundation tarafından hazırlanan rapora göre 2018 Mayısı itibariyle
bilinen vaka sayısı dünyada 30 bireyden azdır.
Belirti ve Semptomlar
Hastalığın az görülmesi ve bireyler arasında farklılık
göstermesi nedeniyle tanı kriterleri kesinleştirilmemiştir. Semptomlar arasında
gelişimde gecikmeler, sakarlık, hipotiroid, koordinasyonda bozukluklar, atak
geçirme, düşük kan şekeri, toksik maddelerin kanda artmasıyla ilgili
rahatsızlıklar ve hastalığın çok geliştiği evrelerde bilinç kaybı yaşanabilir.
Ayrıca, kas dokusunun bozulmasıyla kaslarda ağrı, zayıflık
ve yorgunluk gözükebilir. Doku bozulması, kreatinin kinaz ve miyoglobinin kana
karışmasını sağlayabilir. Bu durumda, miyoglobin böbreklerde birikmeye,
sonrasında da böbreklerin iflas etmesine neden olabilir.
Ayrıca, aritmilere neden olabileceğinden uzun süreli kalp
atışının değişikliği, kalp ve damar hastalıklarına sebebiyet verebilir.
Belirtileri daha da sıralamak mümkündür fakat TANGO2ye bağlı hastalığın olması,
bu belirtilerin kesin gösterileceği anlamına gelmez. Yetkili bir doktora
başvurmak gerekir.
Kalıtım Paterni/Deseni
Otozomal çekinik bir hastalıktır. Eğer bireyin
iki genetic ebeveyni de bu hastalığın taşıyıcısıysa, %25 oranında bu hastalık
kendini gösterebilir. %50 oranında birey, bu hastalığın taşıyıcısı olur ve %25
oranında da hastalıktan etkilenmemiştir/taşıyıcı değildir.
Bununla birlikte, TANGO2 genindeki herhangi bir
mutasyon da farklı bir hastalık veya fonksiyon bozukluğu sağlayabilir.
Araştırmalar devam etmektedir.
Teşhis Yöntemleri ve Tedaviler
Teşhis için yetkili bir doktora ve genetik
teste ihtiyaç vardır. TANGO2 genindeki genetic değişim, bu hastalığın teşhisi
için önemli bir kanıttır. Aile geçmişinde bu hastalık var ise, doğum öncesi
genetic testlere de başvurulabilir.
Hastalığın kontrol altında tutulması ve etkilerinin
azaltılması için metabolic krizler önceden fark edilmeli ve önlem alınmalıdır.
Önlemler kişiden kişiye değişir, genel olarak uzun süreli açlığın ve susuzluğun
önlenmesi gerekir. Tedavi yöntemleri bu hastalığı iyileştirmekten çok
semptomları azaltmayı ve kontrol altında tutmayı amaçlar. Örneğin bir bireyde
başlıca sorun TANGO2 ye bağlı bu hastalığın aritmiye sebep olması ise; aritmi
için tedavi yöntemleri uygulanır.
Hastalıkla İlişkili Genler
TANGO2, taşıma ve golgi aygıtına bağlı bir gendir. Kodladığı proteinin tam işlevi şu anlık bilinmemektedir. Araştırmalar devam etmektedir.
Fotoğraf1. TANGO2 geninin 22. Kromozomdaki yeri
fotoğrafta kırmızı okla belirtilmiştir.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}